Pyridine
La pyridine ou azine, de formule brute C5H5N, est un composé hétérocyclique simple et fondamental qui se rapproche de la structure du benzène où un des groupes CH est remplacé par un atome d’azote. Elle existe sous la forme d’un liquide limpide, légèrement jaunâtre ayant une odeur désagréable et pénétrante (aigre, putride et évoquant le poisson). Elle est très utilisée en chimie de coordination comme ligand et en chimie organique comme réactif et solvant. Les dérivés de la pyridine sont très nombreux dans la pharmacie et dans l’agrochimie. La pyridine est utilisée comme précurseur dans la fabrication d’insecticides, d’herbicides, de médicaments, d’arômes alimentaires, de colorants, d’adhésifs, de peintures, d’explosifs et de désinfectants. Elle est un composé aromatique qui possède une réactivité différente du benzène.
| Pyridine | |
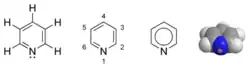
| |
| Structures de la pyridine | |
| Identification | |
|---|---|
| Nom UICPA | Azine |
| Synonymes |
Pyridine, azabenzène |
| No CAS | |
| No ECHA | 100.003.464 |
| No CE | 203-809-9 |
| PubChem | 1049 |
| FEMA | 2966 |
| SMILES | |
| InChI | |
| Apparence | liquide hygroscopique, incolore, d'odeur caractéristique[1] |
| Propriétés chimiques | |
| Formule | C5H5N [Isomères] |
| Masse molaire[2] | 79,0999 ± 0,0046 g/mol C 75,92 %, H 6,37 %, N 17,71 %, |
| pKa | 5,229[3] |
| Moment dipolaire | 2,215 ± 0,010 D[4] |
| Diamètre moléculaire | 0,522 nm[5] |
| Propriétés physiques | |
| T° fusion | −41,15 °C[6] |
| T° ébullition | 115,35 °C[6] |
| Solubilité | dans l'eau : miscible[1] |
| Paramètre de solubilité δ | 21,9 MPa1/2 (25 °C)[7] |
| Masse volumique | 0,98 g cm−3 à 20 °C[8]
|
| T° d'auto-inflammation | 482 °C[1] |
| Point d’éclair | 20 °C (coupelle fermée)[1] |
| Limites d’explosivité dans l’air | 1,7–10,6 %vol[8] |
| Pression de vapeur saturante | 20,5 mbar à 20 °C 35 mbar à 30 °C 95 mbar à 50 °C[8] |
| Viscosité dynamique | 0,95 mPa·s (à 20 °C) |
| Point critique | 56,6 bar à 345,85 °C[6] |
| Thermochimie | |
| S0liquide, 1 bar | 177,9 J K−1 mol−1[6] |
| ΔfH0gaz | 140,2 kJ mol−1[6] |
| ΔfH0liquide | 99,96 kJ mol−1[6] |
| ΔfusH° | 8,278 5 kJ mol−1 à −41,66 °C[6] |
| ΔvapH° | 35,09 kJ mol−1 à 115,25 °C[6] |
| Cp | 133 J K−1 mol−1 (liquide, 25 °C)[6]
|
| PCI | −2 725 kJ mol−1[6] |
| Propriétés électroniques | |
| 1re énergie d'ionisation | 9,25 eV (gaz)[11] |
| Propriétés optiques | |
| Indice de réfraction | 1,507[5] |
| Précautions | |
| SGH[12] | |
  Danger |
|
| SIMDUT[13] | |
  B2, D2B, |
|
| NFPA 704 | |
| Transport[8] | |
| Classification du CIRC | |
| Groupe 3 : Inclassable quant à sa cancérogénicité pour l'Homme[14] | |
| Inhalation | nocif |
| Peau | se laver immédiatement et abondamment avec de l’eau |
| Yeux | idem |
| Ingestion | nocif |
| Écotoxicologie | |
| DL50 | 1,5 g kg−1 (souris, oral) 360 mg kg−1 (rat, i.v.) 1,25 g kg−1 (souris, s.c.) 950 mg kg−1 (souris, i.p.)[3] |
| LogP | 0,65[1] |
| Seuil de l’odorat | bas : 0,23 ppm haut : 1,9 ppm[15] |
| Unités du SI et CNTP, sauf indication contraire. | |










Historique
La pyridine a été découverte en 1851 par le chimiste Thomas Anderson grâce à des études sur la distillation de l’huile d’os et de matières animales. Le mot pyridine provient du grec « pyr » le feu et « idine » est le suffixe utilisé pour les bases aromatiques[16]. Un radical de la molécule est appelé pyridyle. La pyridine ainsi que plusieurs pyridines alkylées ont ainsi été obtenues au début par la pyrolyse des os grâce à une condensation entre l’ammoniac et les aldéhydes ou les cétones, produits par la décomposition du glycérol et des dérivés azotés contenus dans les ossements. La pyridine peut aussi être obtenue par distillation du charbon, du goudron de charbon d’os, le goudron de houille et le goudron à distillation lente, dans les huiles pyrogénées d’origines diverses, (les huiles des schistes bitumineux ainsi que l’huile de café contiennent de la pyridine). La pyridine est ensuite récupérée par lavage de goudron de houille au moyen d’acide sulfurique dilué, la séparation étant ensuite effectuée à l’aide d’alcalins[17]. La structure de la pyridine a été établie en 1869-1870 par Wilhelm Körner et James Dewar[18]. La découverte de la structure de la pyridine a permis le développement de plusieurs voies de synthèse[16] : en 1877, William Ramsay réalise la synthèse de la pyridine à partir d’acétylène et d’acide cyanhydrique. Puis en 1882, Arthur Hantzsch réalise lui aussi une synthèse de la pyridine. Cependant, la pyridine est restée très peu utilisée pendant des décennies et les petites quantités de pyridine utilisées étaient obtenues par distillation du charbon. La pyridine est devenue importante dans les années 1930 avec la découverte de la niacine (vitamine B3), qui prévient les démences. Depuis les années 1940, la 2-vinylpyridine est utilisé dans la synthèse de latex[16]. La demande en pyridine n’a cessé d’augmenter jusqu’à nos jours grâce à la découverte de nombreuses biomolécules pyrimidiques[16].
Synthèse
Synthèse utilisant des aldéhydes ou des cétones avec l'ammoniac
La synthèse à partir d’aldéhydes et/ou de cétones et d’ammoniac est le mode de production de la pyridine le plus courant. L’intérêt de ce type de réaction est l’accès à des réactifs bon marché. La réaction a lieu généralement en phase gazeuse à des températures comprises entre 350 °C et 550 °C en présence d’un catalyseur (composé de silice et d’alumine additionnée d’un métal) et avec un temps de passage dans le réacteur très court, de l’ordre de quelques secondes. Les rendements peuvent atteindre les 60-70 %[16].
La réaction la plus utilisée est la condensation entre l’acétaldéhyde et le formaldéhyde en présence d’ammoniac[19]. Cette réaction se déroule en deux étapes :
- Formation de l’acroléine
- Réaction de l’acroléine avec l’acétaldéhyde en présence d’ammoniac


Le mélange d’acétaldéhyde, de formaldéhyde et d’ammoniac est d’abord préchauffé puis passe dans un réacteur à lit fixe contenant le catalyseur à une température de 400 à 450 °C. Le mélange réactionnel est ensuite refroidi afin de séparer les gaz (hydrogène et ammoniac principalement) des condensats. Une extraction liquide-liquide permet ensuite d’extraire la pyridine et ses dérivés avant qu’une série de colonnes de distillation sépare le solvant d’extraction, la pyridine puis les dérivés plus lourds. Le catalyseur est régulièrement régénéré par le passage d’un flux d’air[19].
Le rendement en pyridine est de l’ordre de 38-63 % en fonction du catalyseur utilisé. Le principal coproduit est la 3-méthylpyridine (rendement entre 9 et 29 %). Si on utilise directement de l’acroléine avec de l’ammoniac, on favorise la synthèse du 3-méthylpyridine avec un rendement de 15-49 %. Et si on utilise uniquement de l’acétaldéhyde avec de l’ammoniac, on produit préférentiellement 2-méthylpyridine et le 4-méthylpyridine avec un rendement de 35-45 % et de 9-44 % respectivement selon le catalyseur utilisé[19].
Déalkylation des alkylpyridines
Les dérivés de la pyridine étant des coproduits facilement vendables, une grande sélectivité n’est pas recherchée durant la synthèse. Toutefois il arrive qu’une part importante des dérivés ayant une valeur trop faible sur le marché soit produite. Dans ce cas, on convertit ces derniers en pyridine par déalkylation oxydative. Les coproduits mélangés avec de l’air ou de l’hydrogène en présence d’eau sont convertis en pyridine avec des rendements compris entre 50 et 93 % en fonction des dérivés et des catalyseurs[19].
Synthèse utilisant des nitriles et l'acétylène
Une autre voie de synthèse est la réaction entre un nitrile et l’acétylène en phase liquide avec un catalyseur au cobalt et permet un rendement d’environ 50 % pour la pyridine[16]. La température de réaction se situe entre 120 °C et 180 °C à une pression comprise entre 0,8 et 2,5 MPa. Cette voie de synthèse est toutefois utilisée pour la production sélective de pyridines ortho-substituées. Ainsi l’acétonitrile et l’acétylène réagissent en présence de cobaltocène pour donner la 2-méthylpyridine avec un rendement de 76 %[19]. L’acrylonitrile réagit avec l’acétylène en présence de (cyclopentadiényl)cobalt-1,5-cyclooctadiène pour donner la 2-vinylpyridine avec un rendement de 93 %[20].
Synthèse de laboratoire
La synthèse de Hantzsch est une autre méthode classique pour obtenir des dérivés de la pyridine. On fait réagir deux équivalents d'un composé 1-3 dicarbonylé (ici l'acétoacétate d'éthyle) avec un équivalent d’ammoniac et un équivalent d’un aldéhyde comme le formaldéhyde pour former une dihydropyridine substituée (cycle ne comportant que deux doubles liaisons). L’action d’un oxydant doux sur cette dihydropyridine permet d’obtenir une pyridine substituée. La synthèse s’effectue à 25 °C et dure plusieurs jours. Les pyridines obtenues sont substituées de façon symétrique.

Un moyen d’obtenir une pyridine dissymétrique est de faire réagir une 3-aminoénone avec un composé 1,3-dicarbonylé. La 3-aminoénone attaque une des fonctions carbonyle du composé 1,3-dicarbonylé pour ensuite donner une imine qui cyclise la molécule en réagissant avec l’autre carbonyle. Au cours de la réaction, deux molécules d’eau sont éliminées[16]. Une pyridine peut être aussi synthétisée à partir d’un composé 1,5-dicarbonylé avec de l’ammoniac pour former une dihydropyridine facilement oxydable en pyridine[16].
Propriétés physico-chimiques
La pyridine est un liquide dans les conditions normales de pression et de température. La pyridine est miscible avec l’eau et avec la plupart des solvants organiques habituels. La pyridine est une molécule de polarité moyenne, moins polaire que l’eau et les alcools mais plus polaire que l’acétate d'éthyle, le dichlorométhane, l’éther de pétrole et les alcanes. En RMN du proton, la pyridine se présente sous trois pics[21] : 8,5 ppm pour les hydrogènes en α de l’azote, 7,6 ppm pour les hydrogènes en γ et 7,2 ppm en β. En RMN du carbone, la pyridine est encore sous trois pics : à 150 ppm pour les carbones 1 et 5, 139 ppm pour le carbone 3 et 123 ppm pour les carbones 2 et 4[21]. En spectroscopie infrarouge, la pyridine présente une bande d’absorption autour de 3 000 cm−1 pour les C-H des carbones sp2[22]. Le seuil de détection olfactif est de 0,02 ppm (dans l’air)[23]. L’indice de réfraction est de 1,510[24]. La constante diélectrique à 25 °C est de 12,4[25].
Aromaticité


La pyridine est un composé aromatique qui vérifie la règle de Hückel. Les électrons délocalisés sont ceux des trois doubles liaisons soit six électrons. Chaque carbone apporte un de ses électrons pi dans la délocalisation et l’azote également hybridé sp2 apporte le sixième électron. La pyridine possède une énergie de résonance de 117 kJ/mol, inférieure à celle du benzène mais supérieure à celles du thiophène, du pyrrole et du furane[26].
Toutes les liaisons carbone-carbone sont de même longueur (139 pm), intermédiaire entre la longueur d'une liaison C-C simple (154 pm) et d'une liaison double C=C (134 pm). Les deux liaisons carbone-azote ont la même longueur (137 pm), plus courte qu'une liaison simple C-N (147 pm) et plus longue qu'une liaison double C=N (128 pm). La pyridine n’est pas un composé absolument plat à cause de la géométrie des liaisons de l’azote. L’atome d’azote possède un doublet électronique libre équatorial, non délocalisé dans le système π aromatique. La paire libre de l’azote est dans une orbitale sp2 dans le plan de la molécule. Seul un électron dans l’orbital p complète l’arrangement électronique de manière à rendre le cycle aromatique.
L’azote ne fait donc pas jouer son caractère mésomère donneur, et seul son caractère inductif attracteur influence le reste du système π. L'atome d’azote exerce donc dans le cycle un effet inductif attracteur et mésomère attracteur. L’azote n’apporte pas de densité électronique supplémentaire. Un effet mésomère attracteur oriente la délocalisation des charges électroniques et affecte une charge électronique sur l’azote dans les quatre formes limites de la pyridine.
L'atome d'azote de la pyridine peut être protoné par réaction avec des acides et forme un cation aromatique appelé ion pyridinium. Le nombre d’électrons délocalisés dans l'ion pyridinium est le même que pour la pyridine, soit six électrons. La charge positive de ce cation est alors stabilisée sur tout le cycle par effet mésomère. L’ion pyridium est isoélectronique au benzène à la différence que l’azote porte une charge. La charge positive diminue les densités électroniques des carbones du cycle notamment pour les carbones qui sont proches de l’azote. Les réactions avec les nucléophiles sont plus faciles avec le pyridinium qu’avec la pyridine, mais les réactions avec les électrophiles sont au contraire rendues plus difficiles. La délocalisation de la charge positive dans le cycle pyrimidique rend le cation plus stable et moins réactif qu'un cation non stabilisé.
Réactivité du noyau pyridinique
Les différents types de réactivités

La pyridine est l’hétérocycle qui a la réactivité la plus proche de celle du benzène. Cependant, la présence d’un atome d’azote dans le cycle déforme la distribution des électrons dans le cycle et la présence d’un doublet libre sur l’azote fournit un site pour la protonation et pour l’alkylation qui n’a aucune analogie vis-à-vis du benzène.
D’après la structure de la pyridine, trois types de réactivités sont attendues[26] :
- la réactivité d’une amine tertiaire : protonation, alkylation, acylation, formation de N-oxyde et coordination avec les acides de Lewis ;
- la réactivité du benzène : réaction de substitution électrophile aromatique, résistance vis-à-vis des additions et des ouvertures du cycle ;
- la réactivité d’une imine conjuguée ou d’un carbonyle : attaque nucléophile en α ou γ.
Le doublet libre de l’azote étant dans une orbitale sp2, il n’est pas délocalisé et confère à la pyridine des propriétés basiques. Cela a pour effet de donner à la pyridine et à ses dérivés des propriétés chimiques assez différentes des dérivés du benzène. L’azote comporte une charge électronique partielle élevée aux dépens des carbones du cycle, qui ont une densité électronique plus faible. En conséquence, la pyridine va réagir plus difficilement que le benzène avec les électrophiles mais plus aisément avec les nucléophiles. Le caractère électroattracteur exercé par l’azote appauvrit considérablement le noyau aromatique.
Les formes mésomères montrent que les positions 2,4 et 6 de la pyridine sont particulièrement appauvries en densité électronique, puisque l'on peut y faire apparaître des charges positives avec les formes de résonance. De plus, l'atome d'azote attirant les électrons du cycle, celui-ci est très désactivé. La pyridine possède du fait de cette attraction un moment dipolaire de 2,2 debye dont le pôle négatif est orienté vers l’azote[26].
En conséquence, les substitutions électrophiles aromatiques telles que la nitration ou l’halogénation se feront plutôt en position 3, mais souvent dans des conditions dures en l’absence de groupements donneurs sur le cycle (amines, éthers…). De même, les alkylations et acylations de Friedel-Crafts sont inconnues sur la pyridine même. En contrepartie, les composés nucléophiles réagissent bien sur le cycle dans les positions appauvries 2, 4 ou 6, en addition ou en substitution.
La pyridine est une imine stable à cause de son aromaticité. En général, les imines sont des intermédiaires de réaction instables[27].
Régiosélectivité des substitutions sur la pyridine
L’azote est un atome très électronégatif qui a une forte tendance à attirer les électrons. Un azote portant une charge négative est donc plus stable qu’un azote avec une charge positive. La position où va s’effectuer une substitution dépend de la stabilité de l’intermédiaire de la réaction. L’écriture des formes mésomères des intermédiaires d’une substitution montre qu’une substitution électrophile aromatique s’effectue préférentiellement en position 3 parce que la présence d’une charge positive sur l’azote est très défavorable tandis qu’une substitution nucléophile aura plutôt lieu en position 2, 4 et 6 parce que la présence d’une charge négative sur l’azote est très favorable.
 Intermédiaires de substitution électrophile en position 2  Intermédiaires de substitution électrophile en position 3 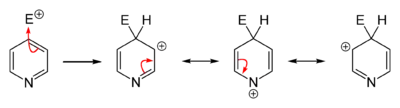 Intermédiaires de substitution électrophile en position 4 |
 Intermédiaires de substitution nucléophile en position 2  Intermédiaires de substitution nucléophile en position 3  Intermédiaires de substitution nucléophile en position 4 |
L'écriture des formes mésomères permet de situer les charges partielles positives de la pyridine en position 2, 4 et 6.

Substitution électrophile aromatique
Les substitution électrophile aromatique sont difficiles à effectuer car la pyridine est moins réactive que le benzène et l’ion pyridinium est encore moins réactif que la pyridine pour les substitutions électrophiles. Lorsqu’une réaction de substitution électrophile a lieu, elle se déroule en position 3 et 5 de la pyridine, qui sont les positions les plus riches en électrons. Le produit cinétique est la pyridine complexée sur le doublet par l’électrophile[26]. La réaction de substitution est difficile car elle doit avoir lieu sur le sel de pyridinium ou bien sur la petite quantité de pyridine non protonée qui est présente et non complexée avec l’électrophile. La présence de substituants donneurs d’électrons va augmenter la facilité de la réaction. Cependant, le passage par un N-oxyde de pyridine est préférable pour faire des réactions de substitutions électrophiles et permet d’obtenir de meilleurs rendements[26].
Les réactions classiques de substitutions électrophiles aromatiques telles que l’alkylation de Friedel-Crafts ou l’acylation de Friedel-Crafts n’ont tout simplement pas lieu. En effet, les acides de Lewis catalysant celles-ci coordonnent l’azote pour le rendre encore plus électroattracteur et les réactions de Friedel-Crafts ont lieu sur l’azote et non sur les carbones de la pyridine[26]. Il est aussi possible d’alkyler ou d’acyler l’azote avec un halogénure d’acide ou d’alkyle sans acide de Lewis et sans autre nucléophile dans le milieu réactionnel. La réaction de Mannich n’a pas lieu avec les pyridines sauf si elles sont substituées par des groupements électrodonneurs[26].
La nitration de la pyridine est difficile et nécessite des conditions fortes. À 370 °C, la pyridine réagit avec l’acide nitrique en présence d’acide sulfurique concentré pour conduire à la 2-nitropyridine (94 %) et à la 3-nitropyridine (6 %). La présence de groupes méthyle sur la pyridine favorise la nitration mais ces groupes sont partiellement oxydés en acide carboxylique. Ces groupes peuvent être éliminés par décarboxylation. Ainsi la luthidine et la collidine permettent d’obtenir la 3-nitropyridine après une nitration et une décarboxylation. De même, la présence d’un groupe hydroxyde ou amine sur la pyridine favorise la nitration. La 4-aminopyridine est nitrée sur la fonction amine puis se transforme en 4-amino-3-hydroxypyridine par réarrangement. La 2-hydroxypyridine est nitrée en position 4. Cependant un autre moyen d’obtenir la 4-nitropyridine est d’utiliser une pyridine N-oxyde avec un rendement de 85 % en présence d’acide sulfurique et d’acide nitrique[26].
À 320 °C, la sulfonation avec de l’oléum conduit à l’acide 3-sulfonique en petite quantité. À 360 °C le produit formé est l'acide 4-sulfonique. La réaction peut avoir lieu à plus basse température avec un catalyseur autour de 220 °C en présence de sulfate mercurique. La pyridine réagit avec les halogènes pour donner des composés cristallisés ou solubles dans le tétrachlorométhane.

Substitution aromatique nucléophile
Généralement, les réactifs nucléophiles effectuent une substitution : d’abord en position 2 et 6 puis en position 4 (bien que certains réactifs fassent l’inverse). L’attaque d’un nucléophile sur la pyridine est suivi de la perte d’un hydrure. La réaction n’est donc pas favorisée thermodynamiquement car l’hydrure est un très mauvais groupe partant et le départ de l’hydrure nécessite des conditions expérimentales vigoureuses. Par contre, si la réaction a lieu sur une pyridine halogénée, la réaction est plus facile car l’ion halogénure est un bon groupe partant. Les 2-halopyridines sont de très bon substrats pour les réactions de substitution nucléophile. Les 4-halopyridines sont des substrats un peu moins bons. Les 3-halopyridines sont très peu réactives[26].
La réaction de Chichibabin est une réaction de substitution nucléophile où un hydrure est remplacé par un groupe amidure. L’action de l’amidure de sodium, de baryum ou de potassium sur la pyridine conduit à la 2-aminopyridine. La réaction peut avoir lieu à sec mais en général, elle s’effectue dans les solvants aromatiques en ébullition. Les groupes hydroxyle, sulfate ou amide en position 2 ou 6 sur la pyridine peuvent aussi être substitués au cours de cette réaction[26]. Dans cette réaction, les 3-alkylpyridines sont substituées en position 2 par un groupe amine tandis que les 2 et 4-alkylpyridine ont du mal à réagir car l’amidure a tendance à arracher un proton de la chaine alkyle pour former un carbanion qui réagit moins bien avec les nucléophiles[26].
La pyridine en présence de soude subit une attaque nucléophile de l'ion hydroxyde pour former la 2-hydroxy-1,2-dihydropyridine qui est oxydée en 2-pyridone avec un faible rendement[26]. L'ion hydroxyde effectue une sustitution nucléophile identique à l'ion amidure mais dans des conditions plus vigoureuses.
La réaction des pyridines avec des alkyllithium ou des aryllithium conduit à des sels de lithium qui peuvent être parfois isolés mais qui en général perdent de l’hydrure de lithium pour être oxydés par le dioxygène et donner des pyridines substituées. La même réaction avec les organomagnésiens est possible mais plus difficile[26].
Oxydation et réduction
Le solvant le plus utilisé pour faire les oxydations en chimie organique est la pyridine. La pyridine est difficilement oxydable et résiste bien aux conditions expérimentales des réactions d’oxydation en milieu basique. Toutefois, elle est oxydée par le permanganate de potassium en présence de potasse (à 100 °C) en libérant du gaz carbonique comme le benzène. La pyridine est aussi oxydée par l’ozone. L’ozonolyse affecte les trois doubles liaisons. Si la pyridine est substituée par des groupements alkyles, ces groupements sont aussi oxydés.
La pyridine traitée par les acides peroxycarboxyliques (le plus souvent l’acide méta-chloroperbenzoïque) ou l’eau oxygénée dans l’acide acétique à 100 °C peut être oxydée en N-oxyde de pyridine. L’action du trichlorure de phosphore ou de la triphénylphosphine sur les N-oxydes de pyridine permet de retourner aux pyridines correspondantes. L’utilisation d’une hydrogénation catalytique douce peut être utilisée pour passer des pyridines N-oxyde aux pyridines simples. Les pyridines N-oxyde sont utilisées dans de nombreuses réactions à cause de leur meilleures réactivité vis-à-vis des substitutions nucléophiles par rapport aux pyridines.
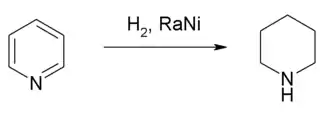
La pyridine est facilement réduite par le tétrahydruroaluminate de lithium LiAlH4 pour former la dihydropyridine puis la tétrahydropyridine et enfin la pipéridine. La pyridine est donc plus facilement réduite que le benzène. L’hydrogénation catalytique de la pyridine à 25 °C, en milieu faiblement acide avec du platine ou faiblement basique avec un amalgame de nickel fournit également la pipéridine[26]. Le borohydrure de sodium n’a pas d’effet sur la pyridine. Cependant, ce réactif réduit les sels de pyridium et les pyridines avec un groupement électroattracteur[26]. La réduction de Birch (sodium en solution dans l’ammoniac) permet de réduire la pyridine. Le radical anion intermédiaire peut se dimériser mais la présence d’une source de proton comme l’éthanol permet d’obtenir une 1,4 dihydropyridine.
Propriétés basiques et réaction sur l'azote
Le doublet de l’azote n’étant pas délocalisé, il confère à la pyridine des propriétés basiques. La pyridine est une base faible (l'ion pyridinium a un pKa de 5,23). Cette faible basicité est en contradiction avec le fait que le doublet semble bien disponible pour capter un proton. L’explication vient de l’hybridation de l’azote. Un composé hybridé sp3 a un effet inductif attracteur plus faible que l’azote hybridé sp2. Le doublet est donc plus lié à l’azote ce qui ne facilite pas sa protonation et diminue sa basicité. La basicité de l’azote permet les mêmes réactions que pour les amines tertiaires.
Les sels de pyridinium sont beaucoup plus réactifs envers les réactifs nucléophiles notamment en position ortho ou para. Ces additions sont parfois suivies d’une ouverture de cycle. Les sels d’alkylpyridine en présence d’hydroxyde d’argent donnent les hydroxydes correspondants et par chauffage en position β ils peuvent perdre une molécule d’eau[26]. La réduction de sel de pyridinium N-acylé par le borohydrure de sodium conduit au 1,2 et 1,4 dihydropyridine[26].
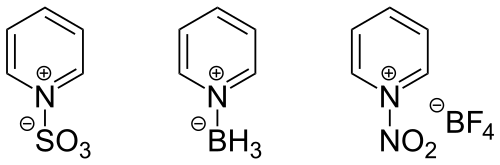
Les acides minéraux et organiques forts forment des sels stables avec la pyridine en protonant l’azote (chlorhydrate et sulfate de pyridium). Ces sels sont très solubles dans l’eau et très hygroscopiques. La présence de groupe à effet inductif donneur facilite la formation de sel en stabilisant la charge positive par effet inductif donneur. Les acides de Lewis sont inhibés par la pyridine car ils sont attaqués par le doublet de l’azote[26].
Les halogénures d’alkyle ou d’aryle activés forment avec la pyridine des sels de N-alkylpyridinium ou N-arylpyridinium. Cette réaction est utilisée pour faire des réactions de deshydrohalogénation qui conduisent aux alcènes. L’acrylonitrile et les acrylates réagissent lors d’une réaction de Mickael pour donner des sels de pyridium[26].
Les chlorures d'acyle réagissent facilement à 0 °C avec la pyridine pour donner des chlorures de 1-acylpyriridinium. Les chlorures d’acide arylsulfonique réagissent de la même manière. Les anhydrides d’acide forment aussi des complexes[26].
Les substituants présents sur la pyridine influencent les propriétés basiques du noyau[28]. Un groupement avec un effet mésomère donneur en position 4 augmente la basicité en stabilisant le cation et en délocalisant la charge positive. La basicité diminue lorsque des groupements avec un effet mésomère attracteur sont sur le cycle.
|
Réaction des chaines latérales
Les pyridines qui portent des groupes méthyle, amine, ou hydroxyle en position 2 ou 4 conduisent en présence de bases fortes à des anions qui sont stabilisés par mésomérie[29]. Les hydrogènes de ces groupes peuvent être plus facilement arrachés et les valeurs de pKa sont donc moins élevées pour ces composés. Lorsque le substituant est en position 3, la stabilisation par mésomérie est moins évidente et par conséquent la valeur du pKa est plus élevée.
|
Utilisation
La pyridine est souvent utilisée comme réactif ou bien comme catalyseur en synthèse organique dans des réactions de condensation, déshalogénation, halogénation ou d’acylation[30] et aussi comme un précurseur pour la synthèse de produits intermédiaires utilisés dans la fabrication d’insecticides, d’herbicides, de médicaments, d’arômes alimentaires, de colorants, d’adhésifs, de peintures, d’explosifs et de désinfectants. La pyridine est alors utilisée comme précurseur à des réactions de substitution nucléophile et plus rarement des substitutions électrophiles ou bien des réactions d’alkylation sur l’azote. La pyridine est aussi utilisée pour dénaturer l’alcool, les antigels et les fongicides, et aussi comme adjuvant pour les teintures textiles.
La pyridine est couramment utilisée comme solvant basique polaire et permet de neutraliser la formation d’acide lors de certaines réactions. La pyridine est souvent utilisée comme solvant polaire aprotique basique ou simplement ajoutée au milieu réactionnel pour neutraliser les acides qui résultent de ces réactions. Cependant la haute température d’ébullition rend parfois la pyridine difficile à éliminer et d’autres solvants organiques avec une faible température d’ébullition sont utilisés.
La pyridine d5 deutérée où les hydrogènes de la pyridine ont été remplacés par des atomes de deutérium peut être utilisée comme solvant en spectroscopie RMN.
La pyridine et ses dérivés peuvent être utilisés pour activer certaines réactions d’acylation ou d’estérification.
- La 4-diméthylaminopyridine (DMAP) est utilisé pour activer les anhydrides lors de réactions d’acylation. L’intermédiaire est un sel 1-acylpyridium qui réagit avec une amine primaire ou secondaire pour former une amide[26].
- La 4-(1-pyrrolidinyl)-pyridine (PPY) permet d’activer une réaction d’estérification entre un acide carboxylique et certains alcools en présence de DCC (dicyclohexylcarbodiimide). Le PPY réagit avec l’intermédiaire formé par la réaction entre l’acide et le DCC, qui se comporte de la même manière qu’un anhydride d'acide[26].
La pyridine est très utilisée comme ligand en chimie de coordination (dans ce cadre, elle est abrégée « py ») car elle a une grande habileté à former des complexes avec de nombreux cations de métaux de transition. La pyridine est un ligand plutôt mou dans la théorie HSAB. Dans les complexes, une liaison azote-métal est formée. Ces complexes peuvent être utilisés pour des analyses sélectives.
Certains des complexes de la pyridine sont utilisés pour oxyder les alcools primaires ou secondaires.
- Le réactif de Collins ou de Sarret sont constitués par un équivalent d’acide chromique et deux équivalents de pyridine. Ils sont préparés par chauffage et diffèrent entre eux par leur forme cristalline.
- Le réactif de Conforth est constitué d’un équivalent de trioxyde de chrome et de deux équivalents de pyridine mélangée avec de l’eau
- Le dichromate de pyridinium PDC est constitué d’un équivalent de dichromate de deux équivalents de pyridine et d’acide chlorhydrique
- Le chlorochromate de pyridinium PCC est constitué d’un équivalent de trioxyde de chrome, d'un équivalent d’acide chlorhydrique et de pyridine.
La pyridine, avec de l’acide barbiturique est couramment utilisée pour la détection colorimétrique des cyanures en solution aqueuse. Elle réagit avec le chlorure de cyanure (formé par la réaction entre l’ion cyanure et la chloramine-T) pour former une espèce conjuguée avec deux molécules d’acide barbiturique, ensemble qui possède une teinte rouge. L’intensité de la coloration est directement proportionnelle à la concentration en cyanure.
Sécurité
Précautions pour la santé

La pyridine est nocive par inhalation, par contact avec la peau et par ingestion[23]. Elle est absorbée par le tractus gastro-intestinal, la peau et les poumons[23]. Les problèmes gastro-intestinaux qui peuvent survenir en même temps sont : des nausées, des vomissements, des anorexies voire des diarrhées[23]. Par inhalation, les vapeurs de pyridine sont irritantes pour les muqueuses oculaires, nasales et respiratoires. Les symptômes habituels d’une exposition à la pyridine sont : maux de têtes, toux, respiration de type asthmatique, laryngite, nausées et vomissement. La pyridine par contact cutané ou par projection provoque une irritation de la peau et des muqueuses, voire des brulures. La pyridine a été à l’origine de quelques cas de sensibilisation cutanée à type d’eczéma. Au niveau oculaire, la pyridine induit des lésions sévères avec opacification cornéenne et cicatrisation de la conjonctive[23]. Les deux principaux organes touchés par la pyridine sont le tractus gastro-intestinal et le système nerveux central. Quelle que soit la quantité absorbée, les problèmes majoritaires sont neurologiques : céphalée, vertiges, asthénie, nervosité, confusion. La pyridine est métabolisée de la même façon pour toutes les espèces étudiées et est éliminée sous forme inchangée ou métabolisée par l’urine. La pyridine n’est pas mutagène.
D'après la fiche de sécurité de l'INRS
| Exposé des risques et mesures de sécurité | |
|---|---|
| R: 11 | Facilement inflammable. |
| R: 20/21/22 | Nocif par inhalation, par contact avec la peau et par ingestion. |
| S: 26 | En cas de contact avec les yeux, laver immédiatement et abondamment avec de l'eau et consulter un spécialiste. |
| S: 28 | Après contact avec la peau, se laver immédiatement et abondamment avec de l'eau. |
| 203-809-9 | Étiquetage CE. |
Précaution pour le stockage

La pyridine dans les conditions normales de température et de pression est un composé stable. Cependant, la pyridine est facilement inflammable et se décompose à température élevée avec des dégagements de vapeurs de cyanure, hautement toxiques[23]. Les vapeurs de pyridine peuvent former dans certaines proportions un mélange explosif avec l’air. Le dioxyde de carbone, les poudres chimiques, les mousses spéciales peuvent être efficaces en cas d’incendie. La pyridine peut attaquer le caoutchouc ou certains plastiques[23]. Pour cela la pyridine doit être stockée dans des récipients en acier ou en fer pour les grandes quantités[23]. Les récipients en verre ne sont utilisés que pour les petites quantités. La pyridine peut réagir vivement avec les acides forts[23] (acide nitrique, acide sulfurique fumant…) ou les oxydants forts[23] (trioxyde de chrome, permanganate de potassium…) et doit être stockée dans des locaux bien ventilés à l’abri de toute source d’ignition, des rayonnements solaires et des sources de chaleurs. Les stockages de pyridine doivent se trouver éloignés des produits oxydants et des acides forts[23].
Effets sur l'environnement
La pyridine est entièrement soluble dans l’eau, et forme des mélanges toxiques même lorsqu’elle est fortement diluée. La pyridine est stable dans l’eau et il ne se produit pas d’hydrolyse donc ce composé reste présent dans les milieux aquatiques pendant une longue période sans se dégrader. La pyridine est une base et fait monter le pH des milieux aquatiques, provoquant des changements importants. Des immissions continues de pyridine dans le milieu aquatique peuvent provoquer une métabolisation accrue de la microflore. Des concentrations de 0,5 mg/l sont suffisantes pour inhiber les processus de nitrification et d’ammonification. Les processus d’oxydation engendrés par la pyridine diminuent sensiblement à partir d’une concentration de 5 mg/l[17]. La pyridine est très mobile dans le sol.
Le rôle de la pyridine comme polluant atmosphérique est mis au jour en 2019. Cette molécule, produite en abondance par l’activité humaine, facilite la formation des aérosols atmosphériques et influence ainsi la formation des nuages et donc le climat[31],[32].
Notes et références
- PYRIDINE, fiche(s) de sécurité du Programme International sur la Sécurité des Substances Chimiques, consultée(s) le 9 mai 2009
- Masse molaire calculée d’après « Atomic weights of the elements 2007 », sur www.chem.qmul.ac.uk.
- (en) « Pyridine », sur ChemIDplus, consulté le 17 septembre 2009
- (en) David R. Lide, Handbook of Chemistry and Physics, CRC, , 89e éd., 2736 p. (ISBN 142006679X et 978-1420066791), p. 9-50
- (en) Y. Marcus, The properties of solvents, Chichester, Angleterre, John Wiley & Sons, coll. « Wiley Series in Solution Chemistry » (no 4), , 254 p. (ISBN 978-0-471-98369-9, présentation en ligne), p. 92
- (en) « Pyridine », sur NIST/WebBook, consulté le 17 septembre 2009
- (en) James E. Mark, Physical Properties of Polymer Handbook, Springer, , 2e éd., 1076 p. (ISBN 978-0-387-69002-5 et 0-387-69002-6, lire en ligne), p. 294
- Entrée « Pyridine » dans la base de données de produits chimiques GESTIS de la IFA (organisme allemand responsable de la sécurité et de la santé au travail) (allemand, anglais), accès le 17 septembre 2009 (JavaScript nécessaire)
- (en) Robert H. Perry et Donald W. Green, Perry's Chemical Engineers' Handbook, USA, McGraw-Hill, , 7e éd., 2400 p. (ISBN 0-07-049841-5), p. 2-50
- (en) Carl L. Yaws, Handbook of Thermodynamic Diagrams : Organic Compounds C8 to C28, vol. 2, Huston, Texas, Gulf Pub. Co., , 396 p. (ISBN 0-88415-858-6)
- (en) David R. Lide, Handbook of Chemistry and Physics, Boca Raton, CRC, , 89e éd., 2736 p. (ISBN 978-1-4200-6679-1), p. 10-205
- Numéro index dans le tableau 3.1 de l'annexe VI du règlement CE N° 1272/2008 (16 décembre 2008)
- « Pyridine » dans la base de données de produits chimiques Reptox de la CSST (organisme québécois responsable de la sécurité et de la santé au travail), consulté le 25 avril 2009
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, « Evaluations Globales de la Cancérogénicité pour l'Homme, Groupe 3 : Inclassables quant à leur cancérogénicité pour l'Homme », sur monographs.iarc.fr, CIRC, (consulté le )
- « Pyridine », sur hazmap.nlm.nih.gov (consulté le )
- (en) Gavin D. Henry, « De novo synthesis of substituted pyridines », Tetrahedron, vol. 60, no 29, (ISSN 0040-4020, DOI 10.1016/j.tet.2004.04.043)
- Ministère fédéral allemand de la Coopération économique et du Développement (BMZ), « Plomb et ses composes organiques », Volume III : Catalogue des normes antipollution, sur gtz.de, Friedr. Vieweg & Sohn Verlagsgesellschaft mbH, Brunswick, (consulté le ).
- (en) Alan J. Rocke, « Koerner, Dewar, and the Structure of Pyridine », B. Hist. Chem., no 2, , p. 4-6 (ISSN 1053-4385, lire en ligne)
- (en) S. Shimizu, N. Watanabe et al., Ullmann's Encyclopedia of Industrial Chemistry, vol. 30, New York, Wiley-VCH, (DOI 10.1002/14356007.a22_399, lire en ligne), « Pyridine and Pyridine Derivatives », p. 557-589
- Brevet DE 2 840 460 H. Bönnemann et M. Samson, π-indenyl-(cycloocta-1,5-diene) cobalt, π-trimethylsilylcyclopentadienyl-(cycloocta-1,5-diene) cobalt, π-cyclopentadienyl-α,α'-bipyridyl cobalt and process for their preparation, and their use, 1980
- (en) Agency of Industrial Science and Technology, « Research Information Database RIO-DB Home Page », sur riodb01.ibase.aist.go.jp, Tsukuba Advanced Computing Center (consulté le ).
- Le spectre est visible sur la page
- Institut national de recherche et de sécurité, « Pyridine », Fiche toxicologique de l’INRS, sur inrs.fr, INRS, (consulté le ), p. 1-6.
- Scientific research 2003/2004, Fluka
- P. Vogel (préf. J.-M. Lehn), Chimie organique : Méthodes et modèles, De Boeck Université, , 1456 p. (ISBN 978-2-8041-2620-9, présentation en ligne)
- R. Milcent et F. Chau, Chimie organique hétérocyclique : Structures fondamentales, chimie et biochimie des principaux composés naturels, EDP Sciences, , 846 p. (ISBN 978-2-86883-583-3, présentation en ligne)
- J. Clayden, N. Greeves et al. (trad. A. Pousse), Chimie organique, De Boeck Université, , 1534 p. (ISBN 978-2-7445-0149-4, présentation en ligne)
- (en) K. Schofield, Hetero-aromatic nitrogen compounds : Pyrroles and pyridines, Plenum Press, , 434 p. (présentation en ligne), p. 146
- (en) Giancarlo Seconi, Colin Eaborn et Alfred Fischer, « Rate constants and solvent isotope effects in the cleavage of picolyl- and (quinolylmethyl)-trimethylsilanes by sodium methoxide in methanol », J. Organomet. Chem., vol. 177, no 1, , p. 129-136 (ISSN 0022-328X, DOI 10.1016/S0022-328X(00)92337-4)
- (en) A. R. Sherman, Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, (ISBN 978-0-470-84289-8, DOI 10.1002/047084289X.rp280, lire en ligne), « Pyridine »
- « Comment une molécule peut changer le climat » [PDF] (consulté le ).
- (en) Linda Feketeová, Paul Bertier, Thibaud Salbaing, Toshiyuki Azuma, Florent Calvo et al., « Impact of a hydrophobic ion on the early stage of atmospheric aerosol formation », PNAS, (DOI 10.1073/pnas.1911136116).
Voir aussi
Bibliographie
- R. Milcent et F. Chau, Chimie organique hétérocyclique : Structures fondamentales, chimie et biochimie des principaux composés naturels, EDP Sciences, , 846 p. (ISBN 978-2-86883-583-3, présentation en ligne)
- J. Clayden, N. Greeves et al. (trad. A. Pousse), Chimie organique, De Boeck Université, , 1534 p. (ISBN 978-2-7445-0149-4, présentation en ligne)
- (en) Erwin Klingsberg et R. A. Abramovitch, Pyridine and its derivatives, Part 1
Articles connexes
Composés structurellement ou chimiquement apparentés :
- Pipéridine, analogue saturé de la pyridine
- Pyrrole, un hétérocyclique aromatique à 4 carbones et un azote
- Bipyridine, un composé polypyridine simple consistant en deux molécules de pyridine liées par une simple liaison
- Diazines : molécules aromatiques avec deux atomes d’azote dans le cycle
- Triazines : molécules aromatiques avec trois atomes d’azote dans le cycle
- 1,2,3-Triazine
- 1,2,4-Triazine
- 1,3,5-Triazine
- noyaux de pyridine et un de benzène fusionnés :
- Aniline, benzène avec un groupe NH2 attaché
- Pyridines substituées
- Picoline, pyridine avec un groupe méthyle
- Lutidine, pyridine avec deux groupes méthyle
- Collidine, pyridine avec trois groupes méthyle
- Acide picolique, pyridine avec groupe COOH en position 2
- Acide nicotinique, pyridine avec groupe COOH en position 3
- Acide isonicotinique, pyridine avec groupe COOH en position 4
- Noyaux aromatiques simples
Liens externes
- (en) « Molecular Orbitals of Pyridine » [« Orbitales moléculaires de la pyridine »] (consulté le )
- Fiches Internationales de Sécurité Chimique Pyridine ICSC : 0323 sur NIOSH
- (en) Synthesis of pyridines (overview of recent methods)
 Portail de la chimie
Portail de la chimie  Portail de la biochimie
Portail de la biochimie