Drépanocytose
La drépanocytose[alpha 1], également appelée anémie falciforme, hémoglobinose S, et autrefois sicklémie, est une maladie génétique résultant d'une mutation sur le gène codant l'hémoglobine. Cette affection peut notamment occasionner un retard du développement de l'enfant, des crises vasoocclusives, une prédisposition aux infections bactériennes et une anémie hémolytique. Elle a été décrite pour la première fois dans la littérature médicale en 1910 par le médecin américain James Herrick et sa transmission héréditaire a été établie en 1949 par James Neel. C'est la maladie génétique la plus fréquente dans le monde, avec environ 310 000 naissances concernées chaque année[1]. Approximativement 80 % des cas de drépanocytose se concentreraient en Afrique subsaharienne[3]. La maladie est également assez fréquente dans certaines régions de l'Inde, de la péninsule arabique et parmi les populations d'origine africaine dispersées de par le monde. Elle a entraîné la mort de 114 000 personnes en 2015[4].
| Drépanocytose Anémie falciforme | ||
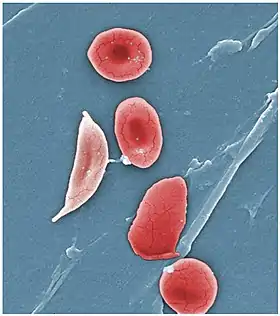 Globule rouge falciforme (pâle, à gauche), typique de la maladie | ||
| Référence MIM | 603903 | |
|---|---|---|
| Transmission | Autosomique récessive | |
| Chromosome | Chromosome 11 p15.5 | |
| Gène | HBB (en) | |
| Empreinte parentale | Non | |
| Mutation | Substitution E6V | |
| Nombre d'allèles pathologiques | 1 | |
| Incidence | 312 000 /an en 2010[1] | |
| Prévalence | 4,4 millions en 2 015[2] | |
| Diagnostic prénatal | Possible | |
| Liste des maladies génétiques à gène identifié | ||
La drépanocytose est due à la substitution d'un seul résidu d'acide aminé sur la chaîne β de l'hémoglobine, protéine des globules rouges assurant le transport de l'oxygène dans le sang. Cette mutation génétique — remplacement du glutamate en position 6 par une valine, donnant de l'hémoglobine S — favorise, dans certaines conditions, l'apparition de globules rouges rigides de forme allongée, dites en faucille ou en feuille d'acanthe, susceptibles d'induire trois grandes catégories de manifestations cliniques qui peuvent être très variables selon les cas : anémie hémolytique chronique avec épisodes d'aggravation aiguë, prédisposition aux infections bactériennes, et manifestations vasoocclusives. Les complications aiguës peuvent notamment revêtir la forme de crises d'anémie aiguës, de séquestrations spléniques, d'infections, d'accidents vasculaires cérébraux (AVC), de syndromes thoraciques aigus ou encore de crises vasoocclusives. Les symptômes apparaissent généralement dès l'âge de 5 à 6 mois et peuvent évoluer vers des douleurs chroniques. L'espérance de vie moyenne des patients dans les pays développés varie entre 40 et 60 ans.
La drépanocytose se manifeste chez les personnes porteuses de deux allèles anormaux du gène HBB, hérités des deux parents. Ce gène est situé sur le chromosome 11. Les personnes n'ayant qu'un seul allèle anormal du gène sont porteuses du trait drépanocytaire et n'expriment pas la maladie. En 2015, on estimait à environ 4,4 millions le nombre total de personnes atteintes de la drépanocytose[2]. Le diagnostic est posé à l'aide d'un test sanguin, certains pays, comme le Royaume-Uni, pratiquant le dépistage pour toutes les naissances. Le diagnostic prénatal est également possible.
Une crise aiguë peut être déclenchée par un changement de température, par un stress, par la déshydratation, ainsi que par une altitude élevée. La prise en charge de la drépanocytose consiste notamment à prévenir les infections à l'aide de vaccins et d'antibiotiques, à assurer une bonne hydratation de l'organisme, à traiter les douleurs induites par les crises, voire en une supplémentation en vitamine B9 (acide folique). On peut également être amené à procéder à une transfusion sanguine ou à l'administration d'hydroxyurée (hydroxycarbamide). Une greffe de moelle osseuse peut permettre la guérison dans un petit nombre de cas.
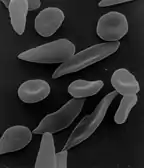
Symptômes
L'affection se signale chez le nourrisson, mais n'est d'ordinaire pas manifeste à la naissance car les globules rouges du nouveau-né contiennent encore de 50 à 90 % d'hémoglobine fœtale. Les symptômes de cette maladie peuvent apparaître dès l'âge de deux à trois mois, lors de l'apparition de la chaîne bêta de l'hémoglobine. Le déclenchement des crises est favorisé par divers facteurs tels qu'une infection, une acidose, une déshydratation, une mauvaise hygiène de vie (tabac, alcool), l'anxiété et le stress, une dépressurisation ou une altitude élevée, un effort physique intense, parmi d'autres causes possibles[5], mais il est bien souvent impossible d'identifier le facteur déclenchant d'une crise particulière. Les crises peuvent être également favorisées par le froid ou un temps sec[6].
La drépanocytose peut conduire à diverses complications aiguës ou chroniques dont plusieurs présentent un taux de mortalité élevé[7].
Manifestations chroniques
Les manifestations chroniques de la drépanocytose associent un retard de taille et de poids, des carences alimentaires (en folates, car cette vitamine est indispensable à la création des hématies qui sont renouvelées très rapidement lors des crises d'anémie, épuisant ainsi le stock de folates), un retard pubertaire fréquent, des troubles cardiopulmonaires (augmentation de la taille du cœur, insuffisance respiratoire), une rate augmentée de volume ou atrophiée, des anomalies rétiniennes (hémorragies), des douleurs chroniques parfois secondaires à des lésions (ulcère de la jambe[8], parfois sans lésion visible, de type neuropathique[9], etc.).
Il peut exister une énurésie[10] probablement favorisée par une hyperfiltration glomérulaire[11].
Crise vasoocclusive
Les globules rouges en forme de faucille tendent à bloquer la circulation sanguine au niveau des capillaires de nombreux organes — os, abdomen, reins, cerveau, rétine, etc. — provoquant douleurs, ischémie, voire nécrose. La fréquence, la durée et la sévérité des crises est extrêmement variable. Les crises douloureuses sont traitées par hydratation, à l'aide d'antalgiques, voire de transfusions sanguines.
La douleur provoquée par les crises légères peut être soulagée à l'aide d'anti-inflammatoires non stéroïdiens (AINS) tels que le diclofénac ou le naproxène. Chez la plupart des patients, les crises sévères requièrent l'injection intraveineuse d'opiacés à intervalles réguliers jusqu'à la fin de la crise. Les crises vasoocclusives affectant des organes tels que le pénis (priapisme[12]) et les poumons sont prises en charge comme des urgences médicales et traitées par transfusion de globules rouges.
Syndrome thoracique aigu
Un syndrome thoracique aigu est caractérisé lorsqu'au moins deux des cinq manifestations suivantes sont observées : douleur thoracique, fièvre, infiltrat pulmonaire, atteinte respiratoire, ou hypoxémie[13]. Ces symptômes étant semblables à ceux d'une pneumonie, il est fréquent de traiter les deux affections en même temps[14]. C'est la deuxième complication la plus fréquente de la drépanocytose, et elle est responsable d'environ 25 % des décès de patients drépanocytaires, la majorité de ces cas présentant également une crise vasoocclusive lorsqu'ils développent un syndrome thoracique aigu[15],[16]. Cependant, environ 80 % des patients développent une crise vasoocclusive au cours d'un syndrome thoracique aigu.
Crise d'asplénie et infections
En raison de ses nombreux capillaires sanguins et de son rôle dans l'élimination des globules rouges défectueux, la rate est fréquemment lésée lors d'une crise drépanocytaire[17]. Elle est généralement atteinte de multiples infarctus tissulaires avant la fin de l'enfance chez les patients homozygotes. L'atteinte de la rate favorise les risques d'infection par des bactéries encapsulées[18],[19], notamment par des pneumocoques[20] ou des méningocoques. Elles peuvent aussi aggraver l'anémie en cas d'infection par le parvovirus B19. Une vaccination préventive, voire la prise d'antibiotiques, peut être recommandée pour se prémunir contre certaines infections particulièrement redoutées chez les patients souffrant d'asplénie fonctionnelle.
Séquestration splénique
La séquestration splénique consiste en une augmentation brutale de la taille de la rate accompagnée d'une chute rapide du taux d'hémoglobine dans le sang. Elle est due à l'accumulation de globules rouges dans la rate aux dépens de la circulation générale. Il s'agit d'une situation d'urgence. Les patients non traités décèdent en une heure ou deux des suites d'un choc hypovolémique. Ces crises sont transitoires, se prolongeant trois ou quatre heures, voire une journée entière[21].
Réticulocytopénie
Les globules rouges des personnes atteintes de drépanocytose ayant une forme anormale sont rapidement éliminés par l'organisme au niveau de la rate, ce qui provoque une anémie hémolytique. Cette anémie peut s'aggraver lors des crises drépanocytaires, ce qui se manifeste par un teint pâle, une accélération de la fréquence cardiaque et un état de fatigue générale. Ces crises sont souvent déclenchées par le parvovirus B19, qui affecte directement la production de globules rouges en détruisant les réticulocytes, qui en sont les précurseurs immédiats. L'infection à parvovirus bloque presque complètement la production de globules rouges pendant deux à trois jours, ce qui est quasiment sans conséquence chez un individu sain, mais peut rapidement menacer la vie d'un patient drépanocytaire. Il faut de quatre jours à une semaine pour se remettre d'une telle crise, certains patients ayant besoin d'une transfusion sanguine pour se rétablir[22].
Crise hémolytique
Une crise hémolytique est une chute brutale de la quantité d'hémoglobine dans le sang en raison de la lyse accélérée des globules rouges. Ce cas est particulièrement fréquent chez les patients atteints de favisme, ou déficit en G6PD[23]. La prise en charge est symptomatique, parfois avec transfusion sanguine.
Autres manifestations aiguës
L'une des manifestations cliniques les plus précoces de la maladie est la dactylite (en), pouvant apparaître chez le nouveau-né âgé d'à peine 6 mois, et susceptible d'être observée chez les enfants porteurs du trait drépanocytaire[24]. La crise peut se prolonger pendant un mois[25].
Complications
On distingue les complications aiguës, qui nécessitent une intervention rapide, et les complications chroniques, qui prennent une part croissante dans la prise en charge de la maladie. Elles débutent dès la petite enfance sous la forme des crises douloureuses vasoocclusives touchant surtout l'os, puis le poumon, la rate et, plus rarement, d'autres organes. La fréquence et le type de ces crises varient selon l'âge, le génotype, et des facteurs environnementaux.
Les autres complications sont les complications infectieuses, celles liées à l'aggravation d'une anémie, et les complications vasculaires. La maladie peut se compliquer d'infarctus cérébraux, dont la prévalence atteint près de 10 % des patients âgés de moins de 20 ans[26]. L'hypertension artérielle pulmonaire est une complication fréquente et grave[27]. Elle est retrouvée en échocardiographie dans près d'un tiers des cas, mais cet examen s'avère peu spécifique, et la prévalence réelle (confirmée par la mesure directe de la pression artérielle pulmonaire par cathétérisme cardiaque) serait bien moindre[28]. La drépanocytose peut également conduire à un tableau d'insuffisance cardiaque à débit élevé, évoluant vers une dysfonction systolique[29].
L'atteinte rénale est fréquente, avec une hématurie ou une protéinurie[30]. L'atteinte oculaire est plus fréquente et plus grave chez les patients SC, combinant l'hémoglobine S avec l'hémoglobine C, par rapport aux patients SS, homozygotes S. Un examen régulier est nécessaire afin de dépister précocement la rétinopathie drépanocytaire qui est de type proliférative, afin de pouvoir proposer un traitement[31].
Durant les années 1980, près de la moitié des malades décédait avant l'âge de 50 ans, au cours d'une crise ou d'un accident vasculaire cérébral[32], avant l'utilisation de l'hydroxyurée[33].
Physiopathologie
Génétique

L'hémoglobine est la protéine des globules rouges assurant le transport de l'oxygène dans le sang. Il s'agit d'un hétérotétramère constitué de deux paires identiques de deux sous-unités de types différents. L'une de ces sous-unités est dite α (alpha) et est codée par un gène situé sur le chromosome 16 tandis que l'autre sous-unité est codée par un gène du chromosome 11 et est soit de type β (bêta), γ (gamma) ou δ (delta), donnant respectivement l'hémoglobine A de formule α2β2 et notée HbA, l'hémoglobine F de formule α2γ2 et notée HbF, et l'hémoglobine A2 de formule α2δ2 et notée HbA2. L'hémoglobine A est de loin la plus abondante des trois, constituant environ 95 % de l'hémoglobine totale d'un adulte sain.
La drépanocytose résulte de la substitution d'une seule base nucléique dans le gène HBB codant la chaîne β de l'hémoglobine A : une paire adénine–thymine est inversée dans la double hélice de l'ADN, ce qui conduit à remplacer un résidu d'adénine par une thymine dans le codon du brin d'ADN sens correspondant au glutamate en position 6, qui se trouve de ce fait remplacé par une valine dans le produit de transcription. Il s'agit d'un polymorphisme nucléotidique.
Brin d'ADN normal antisens 3’ TAC CAC GTA GAC TGA GGA CTC CTC TTC AGA 5’ ||| ||| ||| ||| ||| ||| ||| ||| ||| ||| Brin d'ADN normal sens 5’ ATG GTG CAT CTG ACT CCT GAG GAG AAG TCT 3’ 0 1 2 3 4 5 6 7 8 9 ARN messager normal 5’ AUG GUG CAU CUG ACU CCU GAG GAG AAG UCU 3’ Met-Val-His-Leu-Thr-Pro-Glu-Glu-Lys-Ser-
Brin d'ADN muté antisens 3’ TAC CAC GTA GAC TGA GGA CAC CTC TTC AGA 5’ ||| ||| ||| ||| ||| ||| ||| ||| ||| ||| Brin d'ADN muté sens 5’ ATG GTG CAT CTG ACT CCT GTG GAG AAG TCT 3’ 0 1 2 3 4 5 6 7 8 9 ARN messager muté 5’ AUG GUG CAU CUG ACU CCU GUG GAG AAG UCU 3’ Met-Val-His-Leu-Thr-Pro-Val-Glu-Lys-Ser-
La chaîne β produite ne diffère donc d'une chaîne β normale que par un seul résidu d'acide aminé, en position 6 selon la nomenclature historique, ou 7 selon la nomenclature actuelle[alpha 2]. L'hémoglobine résultante est dite S, initiale du mot anglais sickle signifiant « faucille », et est notée HbS, de formule α2βS2.
Cette mutation s'est probablement produite plusieurs fois de manière indépendante dans des zones géographiques distinctes, comme le suggèrent les études par enzymes de restriction. Ces haplotypes sont dits Sénégal, Bénin, Cameroun, Bantu (originaire d'Afrique centrale) et arabo-indien (péninsule arabique et sous-continent indien). Ils ont une certaine importance clinique, car certains sont associés à une production accrue d'hémoglobine fœtale HbF, notamment Sénégal et arabo-indien, davantage que Bénin et Cameroun, ce qui tend à atténuer les complications de la maladie[34].
Transmission héréditaire et formes composites
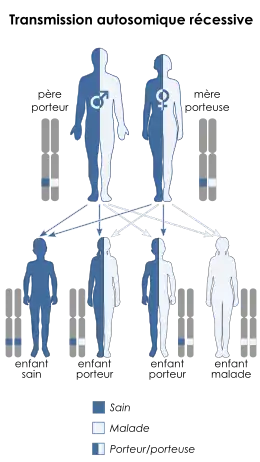
La drépanocytose est une maladie génétique à transmission autosomique récessive. Cela signifie que la mutation affecte un chromosome homologue non sexuel (autosome), et que seuls les homozygotes porteurs de deux allèles mutés (notés SS) sont affectés par la maladie. Les hétérozygotes porteurs d'un allèle muté S et d'un allèle normal A (notés SA) expriment ces deux allèles, car l'allèle S est codominant. On dit de ces personnes qu'elles ont le trait drépanocytaire : elles sont généralement dépourvues des symptômes de la drépanocytose, de sorte qu'on parle de porteurs sains, présentant simplement des hématies falciformes, ou plus ou moins sujettes à la falciformation, qui ne contiennent pas nécessairement un mélange en proportions égales d'HbA et d'HbS.
Compte tenu du caractère autosomique récessif de la transmission de cette maladie, la probabilité de mettre au monde un enfant drépanocytaire est de 100 % si les deux parents sont eux-mêmes atteints de drépanocytose, de 50 % si l'un des deux parents est drépanocytaire tandis que l'autre est porteur du trait drépanocytaire, de 25 % si les deux parents sont porteurs du trait drépanocytaire, et est nulle si l'un des deux parents est sain — et ce même si l'autre parent est drépanocytaire.
Il existe parallèlement des hétérozygotes dits composites, ou doubles, associant la drépanocytose (allèle S) avec une autre hémoglobinopathie, notamment l'hémoglobine C, voire l'hémoglobine E (thalassémie β). La forme SC est la plus courante d'entre elles, formant une hémoglobine HbSC de formule α2βSβC.
Impacts moléculaires et cellulaires
L'hémoglobine S se distingue de l'hémoglobine A par une mobilité électrophorétique plus faible, mais surtout par la solubilité plus faible de sa forme désoxygénée, qui tend à polymériser en chaîne en formant de longues fibres qui déforment le globule rouge. Cela provient de la substitution du résidu de glutamate en position 6 par une valine, c'est-à-dire d'un acide aminé à chaîne latérale chargé négativement par un acide aminé à chaîne latérale aliphatique, et donc apolaire, ce qui crée une zone d'adhérence hydrophobe à la surface de la protéine entre les hélices E et F dans sa forme désoxy, dite tendue.

Ainsi, en situation d'hypoxie — baisse de pression partielle d'oxygène O2 — les molécules d'hémoglobine S tendent à s'agglomérer en longues fibres qui déforment les globules rouges en leur donnant un aspect en faucille ou en feuille d'acanthe. Ce phénomène peut s'observer :
- in vivo, dans le sang veineux et des capillaires, d'où séquestration prolongée, formation de thromboses, et hémolyse facile des globules rouges dans les fins capillaires où les drépanocytes se frayent difficilement un passage, étant donné leur forme allongée, ainsi que dans la rate, où la lenteur de la circulation crée les conditions favorables à la falciformation des drépanocytes, et où ces derniers sont phagocytés par le système réticulo-endothélial.
- in vitro, lors de l'examen du sang frais entre lame et lamelle, avec addition d'un composé réducteur comme le métabisulfite de potassium K2S2O5 ou le métabisulfite de sodium Na2S2O5.
En revanche, le sujet sain présente des globules rouges en forme de disque biconcave quelles que soient les conditions environnementales.
Les globules rouges déformés, ou drépanocytes, sont plus fragiles, ont une durée de vie plus brève, et subissent une destruction anormalement élevée, ce qui conduit à une anémie hémolytique chez les personnes atteintes. La moelle osseuse accélère la production d'érythrocytes mais ne parvient pas à compenser leur destruction lytique.
Pour fixer les idées, un globule rouge fonctionne normalement de trois à quatre mois chez une personne saine, mais les drépanocytes sont détruits au bout de seulement 10 à 20 jours.
Les globules rouges falciformes sont en outre moins souples que les cellules normales, et les épisodes de falciformation répétés tendent à les rendre de plus en plus rigides, de sorte qu'ils conservent leur forme allongée et leur texture rigide même en présence d'une pression partielle d'oxygène élevée. Ces globules rouges sont alors susceptibles de provoquer des ischémies en réduisant l'apport d'oxygène au niveau de différents territoires, d'où des crises douloureuses par infarctus osseux ou infarctus cérébraux, par exemple. Par leur rigidité, les globules rouges falciformes eux-mêmes sont susceptibles d'induire des lésions sur la paroi interne des vaisseaux (endothélium), d'où un risque d'obstruction de ces derniers.
Les hétérozygotes composites SC avec l'hémoglobine C et SE avec l'hémoglobine E (thalassémie β) présentent des syndromes drépanocytaires majeurs (SDM) semblables à ceux des homozygotes SS. L'évolution clinique des composites β-thalassémiques est comparable à celle des patients drépanocytaires. L'hémoglobine composite HbSC, quant à elle, ne polymérise pas comme l'hémoglobine S mais induit la déshydratation du globule rouge, conduisant à sa falciformation. Les enfants porteurs de la mutation SC peuvent présenter les mêmes complications que les enfants homozygotes SS, mais avec une fréquence moindre.
Avantage hétérozygote
La drépanocytose est l'exemple type de maladie offrant un avantage hétérozygote par rapport aux formes les plus graves de paludisme, dues au Plasmodium falciparum : les hétérozygotes SA bénéficient d'une protection estimée entre 60 et 80 % contre ce paludisme, ce qui signifie qu'ils ont une susceptibilité réduite à cette infection[35], et qu'ils présentent des symptômes atténués lorsqu'ils sont touchés[36]. Les homozygotes drépanocytaires SS, en revanche, sont encore plus vulnérables au paludisme que les personnes saines (homozygotes AA), au point que cette maladie est le principal déclencheur de crises drépanocytaires chez ces patients dans les zones impaludées.
Le mécanisme protecteur de l'hémoglobine S chez les hétérozygotes n'est pas entièrement élucidé. De façon générale, cette protection est due à l'interruption du développement du parasite à l'intérieur des globules rouges dans cycle érythrocytaire du parasite (cf. paludisme), en raison de la durée de vie réduite des globules rouges et de leur plus grande fragilité[37]. Le parasite ne peut se reproduire dans ces cellules lorsqu'elles se lysent prématurément, et peuvent difficilement digérer l'hémoglobine S lorsqu'elle est polymérisée, ce qui contribue encore à freiner son développement.
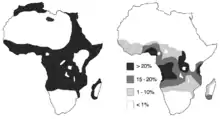
Compte tenu de l'avantage sélectif conféré par l'allèle S du gène HBB dans les zones impaludées, la prévalence de cet allèle y demeure élevée malgré le fort désavantage de la forme homozygote. C'est la raison pour laquelle cette mutation reste fréquente chez les personnes ayant une ascendance récemment issue d'Afrique, du Bassin méditerranéen, d'Inde ou du Moyen-Orient[38] ; le paludisme était également endémique en Europe du Sud jusqu'au milieu du XXe siècle, et n'y est plus présent qu'exceptionnellement, comme en Camargue[39], dans le sud-est de la France.
Examens et diagnostic

L'hémogramme des personnes atteintes de drépanocytose montre une concentration sanguine d'hémoglobine de l'ordre de 6 à 8 g/dL avec un nombre élevé de réticulocytes. Ceci s'explique par le fait que le renouvellement des globules rouges par la moelle osseuse s'accélère afin de compenser l'élimination des cellules falciformes. Le taux d'hémoglobine est plus élevé dans le cas des syndromes drépanocytaires majeurs d'hétérozygotes composites (hétérozygotes SC, SDPunjab, SE, SOArab, etc.). Un frottis sanguin peut montrer des structures caractéristiques d'une asplénie, comme des codocytes et des corps de Howell-Jolly.
La déformation des globules rouges d'un frottis en faucille ou en feuille d'acanthe peut être induite par addition d'un réducteur comme le métabisulfite de potassium K2S2O5, le métabisulfite de sodium Na2S2O5 ou l'acide ascorbique. La présence d'hémoglobine S peut être établie à l'aide d'une solution réductrice, par exemple de dithionite de sodium Na2S2O4, qui se trouble en présence d'HbS mais demeure claire en présence d'hémoglobine normale.
La présence d'hémoglobine anormale peut être détectée par électrophorèse sur gel, un mode d'électrophorèse dans lequel les différentes hémoglobines se déplacent à des vitesses différentes qui permettent de les caractériser. L'hémoglobine falciforme HbS et l'hémoglobine C falciforme HbSC, qui sont les deux principales formes d'hémoglobine drépanocytaire, peuvent être identifiées par cette technique. La chromatographie en phase liquide à haute performance (CLHP) permet de confirmer le diagnostic. Il est rare de devoir recourir à une analyse génétique, car les autres méthodes d'investigation permettent d'identifier précisément l'hémoglobine S et l'hémoglobine C.
Une crise drépanocytaire étant souvent induite par une infection, il est conseillé de rechercher une infection par test urinaire ou radiographie du thorax.
Les personnes ayant le trait drépanocytaire — hétérozygotes SA — peuvent avoir recours à un conseil génétique avant de concevoir un enfant. Le diagnostic prénatal est réalisé à partir d'un échantillon de sang fœtal ou de liquide amniotique, cette dernière technique présentant moins de risques.
Dans les pays développés, hormis les dispositifs de dépistage éventuellement mis en place (comme aux États-Unis, en France ou au Royaume-Uni), le diagnostic se fait en période néonatale si les parents sont malades ou à risque. Dans les pays en voie de développement, le diagnostic se fait souvent à la première manifestation ou complication drépanocytaire ; le dépistage néonatal pourrait se traduire par une amélioration du pronostic[40].
Traitements
Ils comportent un aspect préventif : éviction des facteurs déclenchant les crises (froid, altitude, infections, déshydratation) ; supplémentation en folates (dont l'intérêt réel est cependant aujourd'hui remis en cause), traitement préventif des infections à pneumocoque et méningocoque (vaccination)[41].
Traitements des complications
D'une manière générale, les crises vasoocclusives d'intensité douloureuse modérée, sans fièvre et sans signe de gravité, peuvent être traitées à domicile par hydratation orale abondante (eau alcaline), repos, maintien au chaud et mise sous oxygène. Les antalgiques sont utilisés selon l'intensité de la douleur. La prescription d'opiacés et de morphiniques se fait au mieux en milieu hospitalier[42]. Il est à noter que le métabolisme de la morphine est accéléré chez les sujets porteurs de la maladie, avec une élimination de 3 à 10 fois plus rapide que chez les sujets sains[43].
Les infections sont traitées en milieu hospitalier selon une antibiothérapie couvrant principalement les pneumocoques. La transfusion sanguine est utilisée en cas d'anémie profonde ou d'infection grave, en l'absence de contre-indications. Elle peut être simple ou se combiner en exsanguino-transfusion permettant de réduire la proportion d'hémoglobine S. Ces transfusions sanguines pourraient diminuer sensiblement le risque d'accidents vasculaires cérébraux chez certains enfants particulièrement à risque, tels que ceux présentant une anomalie du doppler transcrânien[44].
La supplémentation en acide folique a longtemps été recommandée par l'OMS mais son efficacité réelle est débattue depuis la fin du siècle dernier[45].
Les patients drépanocytaires ayant une sensibilité accrue au paludisme, on a pu leur recommander, s'ils résident dans une zone impaludée, de prendre une médication prophylactique antipaludéenne[46].
Le traitement de l'ostéonécrose résultant des crises drépanocytaires a pour objectif de réduire les douleurs et de maintenir la mobilité des articulations[47]. Les traitements actuels font appel à la physiothérapie, aux traitements de la douleur, à l'arthroplastie et à la greffe osseuse.
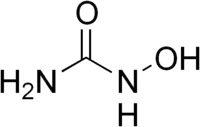
Depuis les années 2000, l'hydroxyurée HONHCONH2, ou hydroxycarbamide, est devenue un traitement de fond. On a montré en 1995 que cette molécule réduisait la fréquence et la gravité des crises[48], et en 2003 qu'elle était susceptible d'allonger l'espérance de vie des patients[49]. L'hydroxyurée permet de stimuler la production d'hémoglobine fœtale[50], formée habituellement en petite quantité et parfaitement fonctionnelle, en remplacement de l'hémoglobine S défectueuse. On a longtemps suspecté une toxicité à long terme, dont on n'a jamais pu montrer qu'elle induisait des risques supérieurs aux bénéfices apportés par son utilisation[7],[33]. Son mode d'action ne permet cependant pas de l'utiliser chez les patients anémiques, ce qui implique une surveillance très soigneuse des paramètres sanguins. L'autre obstacle à son utilisation a d'abord été son coût, qui peut toutefois générer des économies de prise en charge dans les pays développés[51]. Cependant, cette molécule est de plus en plus utilisée dans tous les pays, les études montrant son efficacité et sa bonne tolérance. En 2017, elle est jugée encore sous-utilisée[52].
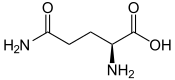
Le 7 juillet 2017, l'usage de la L-glutamine, un acide aminé protéinogène, a été validé aux États-Unis par la FDA[53] chez les patients âgés de 5 ans et plus afin de réduire les complications graves associées à la drépanocytose. Dans un essai clinique de phase 3[54], cette molécule a démontré son intérêt dans la réduction des crises vasoocclusives et dans la diminution de la survenue du syndrome thoracique aigu.
En près de 40 ans (1970-2010), la mortalité infantile des cas de drépanocytose est devenue proche de la population générale, et la durée médiane de vie peut dépasser les 60 ans. Toutefois cette prise en charge est coûteuse, et il existe des inégalités d'accès aux soins selon les pays à haut-revenu[52].
A contrario, certains « médicaments miracles » à l'efficacité non démontrée ont pu être annoncés localement par des personnalités controversées. Ainsi, un remède appelé « VK500 » avait été proposé en 2007 au Bénin contre la drépanocytose ; aucune étude sérieuse n'a cependant jamais trouvé la moindre efficacité thérapeutique à cette préparation[55].
Greffe de moelle osseuse
La greffe de moelle osseuse est potentiellement curative, mais c'est une technique lourde, coûteuse, avec des risques toxiques, et limitée par le manque de donneurs compatibles[52]. En France, la décision est prise lors de réunions de concertation pluridisciplinaires nationales[56].
Les hématies sont produites à partir de cellules souches dans la moelle osseuse. En détruisant la moelle osseuse du malade et en la remplaçant par celle d'un donneur, il y a possibilité d'obtenir une guérison totale. Environ 200 greffes ont été réalisées dans le monde chez des drépanocytaires, permettant d'obtenir la guérison dans 85 % des cas[57]. Il faut cependant un donneur apparenté le plus possible : un frère ou une sœur. Il y a la possibilité pour les parents de recourir à une fécondation in vitro avec sélection par DPI d'embryons compatibles pour la greffe. Cette voie de traitement dite du « bébé médicament » est très encadrée par les lois de bioéthique[58].
Thérapies géniques
Des souris drépanocytaires ont pu être guéries en introduisant chez ces animaux un gène produisant une hémoglobine « anti-drépanocytaire » en quantité élevée[59]. Un essai clinique de phase 1/2 a été programmé à Paris au début du siècle pour évaluer la sécurité et l'efficacité de la thérapie génique des β-hémoglobinopathies (drépanocytose et thalassémie β majeure) par transplantation de cellules souches CD34+ autologues transduites ex vivo avec un vecteur lentiviral β A-T87Q-globine[60].
Un adolescent de 13 ans a ainsi été traité contre la drépanocytose par thérapie génique en octobre 2014[61], et les résultats publiés en mars 2017 se sont révélés très encourageants[62].
Il est cependant peu probable que ces nouvelles thérapies puissent devenir d'utilisation courante à brève échéance en raison de difficultés techniques (validation scientifique), économiques (coût élevé) et éthiques (inégalité d'accès aux soins dans le monde). En attendant, l'extension aux pays en voie de développement des traitements actuellement disponibles dans les pays développés constituerait déjà un progrès significatif. De même, une meilleure compréhension des facteurs environnementaux permettrait de préciser les conseils aux patients atteints de drépanocytose et d'améliorer leur qualité de vie où qu'ils se trouvent[52].
Bonnes pratiques aidant à prévenir les complications
La maladie se manifeste plus ou moins sévèrement selon l'environnement. Un temps froid, une variation de température ou un manque d'oxygène favorisent les complications aiguës, notamment les crises douloureuses. Les complications surviennent plus fréquemment en haute altitude. Dans les pays en voie de développement, les infections sont la principale cause de décès des enfants drépanocytaires, surtout en Afrique. Dans les pays développés, les infections peuvent survenir à tout âge[52].
Une bonne hygiène de vie et des réflexes simples[63] aident à prévenir la survenue des crises et de leurs complications :
- boire fréquemment de l'eau et rester à l'abri des fortes chaleurs (la déshydratation déclenche des crises par augmentation de la viscosité sanguine) ;
- ne pas s'enrhumer, et plus généralement éviter les infections respiratoires ;
- porter des vêtements amples qui ne gênent pas la circulation sanguine, éviter les positions contraignantes (jambes croisées) ;
- éviter l'essoufflement, les endroits mal aérés (voyages en avion, plongée sous-marine) et les altitudes élevées (supérieures à 1 500 m).
Épidémiologie, écoépidémiologie
La drépanocytose est la maladie génétique monogénique (liée à un seul gène) la plus répandue dans le monde[52]. Selon l'OMS, 300 000 enfants naissent dans le monde chaque année avec une hémoglobinopathie grave, dont 200 000 enfants atteints de drépanocytose en Afrique[64]. En 2015, la grande majorité de ces naissances a eu lieu dans trois pays : Nigeria, République démocratique du Congo, Inde[52]. Le nombre de porteurs sains de thalassémies est en réalité plus élevé que celui du gène drépanocytaire, mais la prévalence de ces derniers dans certaines régions du monde conduit localement à un plus grand nombre d'homozygotes atteints par la maladie[64].
L'allèle S, responsable de l'anomalie, est surtout répandu dans le continent africain (atteignant dans certaines populations la fréquence de 30 %) ; on le trouve également en Inde, en Arabie saoudite et dans d'autres régions du bord de la Méditerranée, en Italie (surtout en Sicile), en Grèce et en Anatolie. Les migrations ont accru la fréquence de cette maladie sur le continent américain.
Cette distribution se superpose assez bien avec celle d'une autre maladie d'origine infectieuse, le paludisme, ou malaria, notamment la forme à Plasmodium falciparum, la plus grave. La présence élevée de la drépanocytose en Afrique semble être un cas de polymorphisme génétique équilibré résultant d'une sélection naturelle par avantage hétérozygote : en effet, les porteurs sains — hétérozygotes SA, ayant le trait drépanocytaire — sont relativement protégées contre P. falciparum. La preuve épidémiologique de cette protection a été apportée par plusieurs études depuis 2002[37].
L'apparition, puis la sélection, de cette mutation se sont faites en différents points du monde, coïncidant avec les zones d'endémie du paludisme. Ces événements se seraient produits entre 1 100 et 200 av. J.-C. en plusieurs endroits d'Afrique et d'Asie, selon Tchernia[65] ; ou il y a environ 70 000 à 150 000 ans, selon Bartolucci, qui précise que, parmi les différents haplotypes connus (ensembles de gènes liés), cinq seraient apparus de façon indépendante : haplotype Sénégal (côte atlantique de l'Afrique de l'Ouest), haplotype Bénin (golfe du Bénin), haplotype Cameroun, haplotype Bantu (Afrique équatoriale), haplotype arabo-indien (sous-continent indien et péninsule arabique)[37].
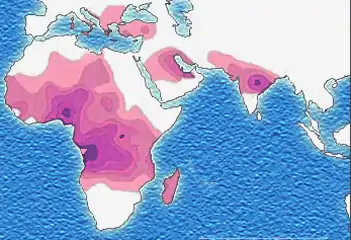 Distribution historique du gène drépanocytaire.
Distribution historique du gène drépanocytaire.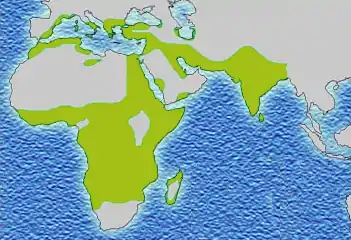
Certains cervidés — plusieurs espèces de muntjacs — sont également porteurs d'une mutation proche de celle de la drépanocytose, anomalie connue depuis 1840 mais dont la cause génétique n'a été publiée qu'en 2017[66]. Chez ces animaux, la mutation consiste, comme chez l'homme, en la substitution de l'acide aminé glutamate par une valine, mais pas au même emplacement que dans l'hémoglobine humaine. Contrairement à ce que l'on observe chez les humains, ces cerfs produisant des globules rouges falciformes semblent en parfaite santé[66]. De plus, la falciformation survient chez l'homme lorsque les globules rouges sont soumis à une faible pression partielle d'oxygène, alors qu'elle survient lorsque la pression partielle d'oxygène est élevée chez le muntjac[66].
Afrique
Dans certaines parties de l'Afrique subsaharienne, la drépanocytose touche jusqu’à 2 % des nouveau-nés. La fréquence du trait drépanocytaire, c'est-à-dire le pourcentage de porteurs sains qui n’ont hérité du gène mutant que d’un seul des parents, atteint 10 à 40 % en Afrique équatoriale, 1 à 2 % sur la côte de l’Afrique du Nord et moins de 1 % en Afrique du Sud.
Dans les pays d’Afrique de l'Ouest, comme le Ghana et le Nigeria, la fréquence du trait drépanocytaire atteint 15 à 30 %. En Ouganda, cette fréquence atteint 45 % chez les Ambas[64]. Au Nigeria, pays le plus peuplé d'Afrique, le gène de l'hémoglobine S est présent chez 24 % de la population, de sorte que ce sont pas moins de 150 000 enfants drépanocytaires qui naissent chaque année dans ce seul pays[64].
Dans ces pays africains, la mortalité des enfants de moins de 5 ans atteints de drépanocytose peut atteindre 90 % (malnutrition et pauvreté, manque de dépistage et de vaccinations), alors que la mortalité infantile toutes causes confondues a globalement été réduite dans l'ensemble des pays en voie de développement depuis les années 1990-2010[52].
Moyen-Orient
En Arabie saoudite, environ 4,2 % de la population est porteuse du trait drépanocytaire et 0,26 % est atteint par la maladie. La prévalence la plus élevée se trouve dans la province d'ash-Sharqiyah, où elle atteint 17 % pour le trait drépanocytaire et 1,2 % pour la drépanocytose[67]. Un programme de dépistage prénuptial obligatoire par électrophorèse avec conseil génétique a été institué en 2005 dans tout le royaume afin de réduire l'incidence la drépanocytose et des thalassémies[68].
À Bahreïn, une étude publiée en 1998 couvrant 56 198 personnes hospitalisées a établi que 2 % des nouveau-nés étaient atteints par la maladie, 18 % étaient porteurs du trait drépanocytaire et 24 % d'un gène de thalassémie β[69]. Les femmes enceintes ont commencé à être systématiquement dépistées en 1992, ainsi que les nouveau-nés si leur mère était porteuse du gène. Un conseil prénuptial gratuit a été institué en 2004. Ces mesures ont été appuyées par un programme d'information du public[70].
Inde et Népal
La drépanocytose est fréquente chez certains peuples d'Inde centrale, au sein desquels la prévalence de la maladie varie de 9,4 % à 22,2 % dans les régions endémiques du Rajasthan, du Madhya Pradesh et du Chhattisgarh[71]. Elle est également endémique chez les Tharus du Népal et d'Inde, qui présentent une incidence réduite du paludisme bien que vivant dans des régions impaludées.
Amériques
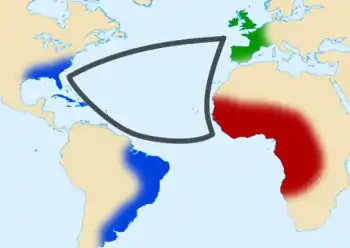
La traite des esclaves africains a introduit la drépanocytose sur le continent américain, notamment aux États-Unis, au Brésil et dans les Caraïbes[72]. L'hémoglobine S est absente chez les Amérindiens, les populations originaires d'Europe du Nord et d'Océanie, alors qu'elle est relativement fréquente chez les Afro-Américains[72].
Aux États-Unis, le nombre de malades était estimé en 2016 à environ 100 000 personnes, le gène drépanocytaire étant présent chez environ 1⁄13 des Afro-Américains[73], ce qui correspond à environ 7,7 %. Une étude de 1985 avait trouvé un taux analogue, de l'ordre de 5 à 6 %, chez les Noirs du Brésil, ainsi qu'une prévalence voisine de 1 % chez les Brésiliens identifiés comme Blancs[74].
La maladie est absente en Alaska, ainsi que dans certains pays d'Amérique du Sud le long de la façade de l'océan Pacifique (Équateur, Pérou, Bolivie, Chili). La situation est intermédiaire dans le reste du continent (Canada, Amérique centrale, Argentine)[52].
Europe
Dans plusieurs pays ou régions d'Europe (sud de l'Italie, Grèce, Albanie), la drépanocytose est une maladie autochtone, avec des fréquences de porteurs du trait drépanocytaire entre 1 et 5 % de la population.
Dans d’autres pays européens (Royaume-Uni, France métropolitaine, Belgique, Allemagne), les flux migratoires, à partir des années 1960, en provenance d'Afrique, du Moyen-Orient et d'Asie du Sud, ont été à l'origine d'un accroissement des cas diagnostiqués, variable selon les spécificités historiques de chaque pays[75].
Dans la péninsule ibérique, les cas de drépanocytose majeure étaient encore rares en Espagne dans les années 2000, mais tendent à augmenter avec l'immigration récente. Au Portugal, dont l'histoire coloniale est plus ancienne, les études génétiques ont décelé deux vagues d'immigration apportant la mutation drépanocytaire : l'une entre les VIIIe siècle et XIIIe siècle venant de Méditerranée, une autre à partir du XVe siècle venant d'Afrique. La mutation s'est diluée dans l'ensemble de la population portugaise actuelle[75].
En Allemagne, le nombre de cas de drépanocytose n'est pas suivi de manière exhaustive. Seuls 300 cas de syndromes drépanocytaires majeurs étaient recensés en 1997[76], tandis qu'une étude de 2017 portant sur les personnes arrivées dans le pays à la suite de la crise migratoire en Europe estimait à 3 000 le nombre de malades drépanocytaires au sein de cette population, précisant que le nombre de patients de nationalité allemande n'est pas connu[77]. Au Royaume-Uni, le Département de la Santé estimait en 2009 à environ 12 500 le nombre de cas de drépanocytoses dans la seule Angleterre[78].
France
Les premiers cas de drépanocytose ont été rapportés en métropole dans les années 1940[79]. Jusqu'aux années 1950, ces cas concernaient presque uniquement des personnes d'origine antillaise[75]. Le premier mouvement migratoire fin XIXe siècle - début XXe siècle en provenance d'Europe du Sud n'a pu être dépisté.
Après la décolonisation des années 1950-1960 survint un deuxième mouvement migratoire provenant d'Afrique et d'Asie du Sud-Est. Des études ponctuelles de dépistage furent alors menées à partir des années 1970 dans plusieurs régions. Entre autres, en 1977, un dépistage systématique de la population scolaire de la région marseillaise sur plus de 35 000 élèves, a permis d'établir que 0,22 % étaient porteurs du trait drépanocytaire, tous issus de familles originaires de pays à risques, certains Italiens du sud vivant en France depuis cinq générations[79].
En l'absence de registre national sur la drépanocytose en France — comme d'ailleurs dans les autres pays développés ayant mis en place un dépistage néonatal systématique, tels que le Royaume-Uni et les États-Unis — on ignore la prévalence de cette maladie dans la population de ces pays. Selon une évaluation de 2004, la France comptait quelque 6 000 patients drépanocytaires, c'est-à-dire homozygotes SS et hétérozygotes composites avec l'hémoglobine C ou la thalassémie β à hémoglobine E[80]. C'est la plus fréquente des maladies génétiques repérées par dépistage néonatal : en 2016, 356 nouveau-nés ont été confirmés positifs à la drépanocytose en France métropolitaine, contre 315 cas d'hypothyroïdie congénitale et 130 cas de mucoviscidose, par exemple[81].
Dépistage
Les méthodes permettant un dépistage néonatal systématique sont disponibles depuis les années 1970. Toutefois, leur mise en œuvre ne s'est généralisée qu'une quinzaine d'années plus tard, d'abord aux États-Unis à partir de 1987, puis au Royaume-Uni. La raison principale de ce décalage est que, en l'absence de traitement efficace, on pensait qu'un diagnostic précoce n'aurait aucun effet sur les taux de morbidité et de mortalité de la maladie[82]. Cependant, dans quelques établissements où le dépistage était associé à une prise en charge médicale avec information des parents, des études finirent par mettre en évidence une réduction impressionnante des infections et de la mortalité des enfants drépanocytaires, notamment grâce à une antibiothérapie prophylactique adaptée avant l'âge de 4 mois. Une étude avec groupe placebo, prévue pour une surveillance de deux ans, fut interrompue huit mois avant son terme car les résultats étaient déjà significatifs[82]. En 1987, à Bethesda, aux États-Unis, une conférence de consensus médicale conclut à la nécessité, aux États-Unis, d'un dépistage généralisé et systématique à tous les enfants, avec, pour les parents, la liberté de refuser. Ce dépistage devait s'effectuer sous le contrôle de centres spécialisés avec prise en charge médico-sociale : médecins, travailleurs sociaux, information optimale avec conseil génétique et conseil de prévention[82].
Cette nouvelle approche de la prise en charge fut rapidement adoptée par le Royaume-Uni, qui disposait, en 1992, de 16 centres, dont quatre dans la région de Londres. À cette date, la France métropolitaine accusait quelques années de retard, tandis qu'un programme systématique de dépistage et de prise en charge existait en Guadeloupe depuis 1990[83] ; aujourd'hui, contrairement à d'autres pays (États-Unis et Royaume-Uni notamment) et à d'autres maladies génétiques, comme la phénylcétonurie et la mucoviscidose, le dépistage néonatal de la drépanocytose n'est pas systématique en France métropolitaine[84] ; il l'est en revanche dans les départements et régions d'outre-mer : Guadeloupe, Martinique, Guyane, Mayotte et La Réunion.
Dans les pays en voie de développement, le dépistage de la maladie n'est pas systématique, et est généralement réalisé à l'issue d'une première crise, ce qui peut retarder le résultat de plusieurs mois dans la mesure où l'urgence est avant tout de traiter l'anémie sévère et les complications de la crise pour lesquelles l'enfant a été amené à consulter. Une méthode rapide et peu coûteuse a été testée avec succès début 2017 au Togo, au Mali et en République Démocratique du Congo[85] ; elle permet d'avoir un résultat en 30 minutes pour toute personne âgée d'un an ou plus en laissant tomber du sang du patient sur un buvard : l'hémoglobine A s'étale sur le buvard en formant une auréole claire, l'hémoglobine S reste au centre en formant une tache rouge, tandis que les hétérozygotes présentent une juxtaposition de ces deux motifs[86]. Ce dépistage n'est cependant utile que dans les zones où sont possibles des interventions préventives et thérapeutiques prenant en charge les personnes dépistées[52].
Histoire
Élucidation de la maladie

La première description moderne de la drépanocytose remonte peut-être à 1846 avec la publication aux États-Unis de l'autopsie d'un esclave fugitif dépourvu de rate[87],[88]. Des cas d'esclaves montrant une résistance au paludisme et ayant tendance aux ulcères de la jambe ont également été décrits[88]. En 1874, un médecin africain de l'armée britannique originaire de Freetown, « James Beale » Africanus Horton (en), décrivit une rhumatic fever, équivalent clinique de la drépanocytose, correspondant aujourd'hui à une crise vasoocclusive osseuse, mais ce travail passa inaperçu[89].

Les caractéristiques anormales des globules rouges ont été décrites pour la première fois en 1910 par Ernest Irons (en) et James Herrick à partir du cas de Walter Clement Noel, un étudiant en odontologie d'une vingtaine d'années originaire de la Grenade, dans les Antilles : ce patient était traité à Chicago pour une anémie depuis 1904[90],[91], puis pour des « rhumatismes musculaires » et des « attaques biliaires », avant de mourir d'une pneumonie à Saint-Georges en 1916[90],[92]. Ce patient souffrait de toux, de fièvre, de vertiges, de maux de tête et d'un état de faiblesse, et ressentait des palpitations et un essoufflement, comme certains membres de sa famille. Son sang montrait qu'il était très anémique, le nombre de ses globules rouges n'atteignant que la moitié de la valeur normale. L'observation d'un frottis sanguin montra des globules rouges de forme inhabituelle en faucille — c'est-à-dire « falciforme » — ou en feuille d'acanthe, résultat publié en novembre 1910[93]. Quelques mois après cette publication, un autre article intitulé exactement de la même manière — Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia[94] — décrivit le cas d'un patient admis à l'hôpital de l'université de Virginie le 15 novembre 1910 ; la publication qu'en fit Verne Mason (en) en 1922 employait pour la première fois le terme « anémie falciforme » pour définir cette maladie[92],[95].

En 1917, Victor E. Emmel parvint à reproduire la falciformation in vitro chez certains sujets cliniquement sains, et conclut à l'existence de deux formes de la maladie. Par la suite, des études familiales envisagèrent l'hypothèse d'une transmission héréditaire autosomique récessive avec des formes manifestes et des formes latentes ou silencieuses. Les facteurs de falciformation furent précisés, dont la pression partielle d'oxygène, ainsi que la durée de vie plus courte des globules rouges falciformes[96]. C'est en 1933 que furent ainsi décrits la drépanocytose d'une part, et le trait drépanocytaire d'autre part, grâce aux travaux de Lemuel Diggs (en), mais il fallut attendre 1949 pour que James Neel (en) établisse les propriétés génétiques de la maladie et l'existence d'une forme homozygote héritée de parents hétérozygotes[92],[97]. C'est également en 1949 que les Américains Linus Pauling et Harvey Itano et al. décrivirent la solubilité abormale de l'hémoglobine S et attribuèrent ces anomalies à la molécule d'hémoglobine elle-même, la discrimination entre hémoglobine S et hémoglobine A étant réalisée par électrophorèse des protéines[92],[98] ; ce fut la première description de la base moléculaire d'une maladie génétique.
La compréhension des liens entre paludisme et drépanocytose s'affina entre la fin des années 1940 et le début des années 1950, tandis que la nature précise de l'altération moléculaire conduisant à l'hémoglobine S — mutation d'un acide aminé sur la chaîne β de l'hémoglobine — fut précisée par le Britannique Vernon Ingram (en) en 1956[99], et que l'affinage des techniques d'électrophorèse de l'hémoglobine permit en 1954 d'identifier d'autres hémoglobinopathies, comme celle à hémoglobine HbSC, combinant hémoglobine S et hémoglobine C[92]. Cela démontra pour la première fois que les gènes déterminent la nature de chaque acide aminé dans une protéine.
En 1978, Tom Maniatis isola le gène HBB de la chaîne bêta de l'hémoglobine sur le chromosome 11.
En 1980, Yuet Wai Kan (en) met au point un test génétique prénatal de la drépanocytose[100].
La preuve épidémiologique de la protection de la drépanocytose contre le paludisme à P. falciparum fut apportée en 2002, confirmant ainsi l'hypothèse de Haldane en 1949 qui s'appuyait sur des superpositions de cartes géographiques (fréquence de la drépanocytose et du paludisme). La présence des gènes de la drépanocytose peut donc s'expliquer par une pression de sélection induite par le paludisme. C'est un avantage hétérozygote contre le paludisme, mais au risque de maladie sévère dans sa forme homozygote[37].
En Afrique
La drépanocytose aurait été connue des populations africaines, notamment au Ghana. L'enfant atteint est perçu comme un être réincarné, bénéfique ou maléfique selon les régions, un être de passage qu'il faut essayer de retenir, ou comme revenant sans cesse pour tourmenter ses parents[89]. La maladie est perçue comme transmise par la mère, notamment dans les sociétés polygames : si l'homme et l'une de ses femmes ont chacun le trait drépanocytaire, un quart de leurs enfants seront homozygotes, et donc atteints de drépanocytose, en vertu de la transmission autosomique récessive de la maladie, ce qui peut aboutir à des répudiations[65].
En Occident
Identifiée aux États-Unis (4 cas décrits jusqu'en 1922), la maladie fut d'emblée qualifiée de maladie des Noirs, la cause restant inconnue. La recherche sur la maladie demeura exclusivement américaine jusque dans les années 1940. Quand la maladie était détectée chez quelques Blancs, elle s'explique par une erreur de diagnostic ou un métissage lointain. Les médecins coloniaux européens considèrent d'abord que la maladie devait être rare en Afrique, et que son importance en Amérique est due à l'environnement ou au métissage américain plus fréquent. Sa grande fréquence en Afrique n'est reconnue que progressivement[89].
Le trait drépanocytaire — forme hétérozygote de la maladie — est alors utilisé comme traceur de migration et moyen d'étude sur les origines de l'humanité[101]. Son existence indiquerait la présence d'une population originelle plus primitive. Ainsi, sa découverte en Inde centrale dans des populations à peau noire a conduit à l'hypothèse d'une origine indienne des populations africaines[89].
Le concept de race humaine fut largement remis en cause dans la seconde moitié du XXe siècle, tandis qu'on trouvait toujours davantage d'hémoglobine S au sein de populations blanches, fait peu compatible avec la conception de la drépanocytose comme « maladie des Noirs ». À partir des années 1960-1970, la recherche génétique montra que la maladie est due à une mutation indépendante des autres gènes[89]. La présence de l'hémoglobine S est donc indépendante de la couleur de la peau, et on ne considère plus quatre ou cinq grandes races humaines, mais des milliers de gènes plus ou moins indépendants, dont celui de la drépanocytose.
Prise en charge médico-sociale
Dans les années 1960, aux États-Unis, l'information des familles et la prise en charge médicale des enfants drépanocytaires sont quasi clandestins et le fait de sociétés de bienfaisance ou de mouvements politiques noirs, comme les Black Muslims de Malcolm X ou le mouvement des droits civiques de Martin Luther King. Des extrémistes noirs prétendirent que la maladie était une invention des Blancs pour promouvoir un génocide, tandis que Linus Pauling proposait de tatouer les hétérozygotes pour mieux interdire leur union. Les porteurs sains (hétérozygotes porteurs du trait drépanocytaire) étaient stigmatisés, avec refus d'emploi, de prêt bancaire ou d'assurance, par exemple[65].
En 1971, le président Richard Nixon promit des crédits pour la recherche et la prise en charge des malades. En 1972, il signe une nouvelle loi the Sickle Cell Anemia Control Act. Dans les années qui suivirent, on observa une corrélation directe entre les crédits alloués par le National Institutes of Health[alpha 3] (NIH) et l'amélioration de la qualité et de la durée de vie des drépanocytaires américains[65].
Cependant, dans les années 1980, des patients reçus en urgence pour crise douloureuse drépanocytaire sont encore considérés aux États-Unis comme des toxicomanes en état de manque, car ils connaissent mieux que les soignants les opiacés qui les soulagent au mieux[102].
Dans les années 1990, dans d'autres pays développés, comme en Grande-Bretagne ou en France, il se confirme finalement que la drépanocytose est une maladie génétique dont le pronostic dépend directement de la qualité de la prise en charge médico-sociale[103].
Dans les années 2010, la drépanocytose reste un défi social aux États-Unis : les patients drépanocytaires doivent « être reçus et traités comme des personnes ayant besoin d'un soulagement, et qui doivent être soutenues plutôt que stigmatisées dans une ambiance électrique »[102].
Filmographie
- L'amour fleurit en décembre (A Warm December, 1973), histoire d'amour entre un médecin (incarné par Sidney Poitier), et une patiente drépanocytaire[102].
- Le Doigt sur la plaie, film documentaire burkinabé d'Abdoulaye Dao (2008).
- Les lances de Sickle Cell ou chroniques d'une douleur annoncée (2009).
Malades célèbres
- Le rappeur Prodigy, du duo Mobb Deep, avait été diagnostiqué à l'âge de trois mois[104].
- Le joueur de tennis Jo-Wilfried Tsonga, tardivement diagnostiqué[105].
- L'acteur Bolaji Badejo, qui joue l'alien dans le film Alien de Ridley Scott[106].
Notes et références
Notes
- Du grec ancien δρέπανον / drepanon « faux », « serpe », employé ici dans le sens de faucille, et κύτος / kutos « enveloppe », « objet creux », employé dans le sens de cellule vivante.
- La littérature consacrée à la drépanocytose remonte jusqu'aux années 1940, et on a longtemps ignoré le résidu de méthionine initial dans la numérotation des acides aminés des sous-unités de l'hémoglobine, de sorte qu'il est généralement fait mention d'une substitution E6V pour définir cette mutation ; la préconisation actuelle est cependant de commencer la numérotation en incluant la méthionine initiale, ce qui conduit à définir la mutation de l'hémoglobine S plutôt comme une substitution E7V.
- Équivalent de l'INSERM en France.
Références
- (en) Frédéric B. Piel, Anand P. Patil, Rosalind E. Howes, Oscar A. Nyangiri, Peter W. Gething, Mewahyu Dewi, William H. Temperley, Thomas N. Williams, David J. Weatherall et Simon I. Hay, « Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates », The Lancet, vol. 381, no 9861, , p. 142-151 (PMID 23103089, PMCID 3547249, DOI 10.1016/S0140-6736(12)61229-X, lire en ligne)
- (en) GBD 2015 Disease and Injury Incidence and Prevalence Collaborators, « Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015 », The Lancet, vol. 388, no 10053, , p. 1545-1602 (PMCID 5055577, DOI 10.1016/S0140-6736(16)31678-6, lire en ligne)
- (en) David C. Rees, Thomas N. Williams et Mark T. Gladwin, « Sickle-cell disease », The Lancet, vol. 376, no 9757, , p. 2018-2031 (PMID 21131035, DOI 10.1016/S0140-6736(10)61029-X, lire en ligne)
- (en) GBD 2015 Mortality and Causes of Death Collaborators, « Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015 », The Lancet, vol. 388, no 10053, , p. 1459-1544 (PMID 27733281, PMCID 5388903, DOI 10.1016/S0140-6736(16)31012-1, lire en ligne)
- (en) N. A. Alli, M. Patel, H. D. Alli, F. Bassa, M. J. Coetzee, A. Davidson, M. R. Essop, A. Lakha, V. J. Louw, N. Novitzky, V. Philip, J. E. Poole et R. D. Wainwright, « Recommendations for the management of sickle cell disease in South Africa », The South African Medical Journal, vol. 104, no 11, , p. 743-751 (PMID 25909112, DOI 10.7196/SAMJ.8470, lire en ligne)
- (en) Simon Jones, Edward R. Duncan, Nikki Thomas, Joan Walters, Moira C. Dick, Susan E. Height, Adrian D. Stephens, Swee Lay Thein et David C. Rees, « Windy weather and low humidity are associated with an increased number of hospital admissions for acute pain and sickle cell disease in an urban environment with a maritime temperate climate », British Journal of Haematology, vol. 131, no 4, , p. 530-533 (PMID 16281945, DOI 10.1111/j.1365-2141.2005.05799.x, lire en ligne)
- (en) Barbara P. Yawn, George R. Buchanan, Araba N. Afenyi-Annan, Samir K. Ballas, Kathryn L. Hassell, Andra H. James, Lanetta Jordan, Sophie M. Lanzkron, Richard Lottenberg, William J. Savage, Paula J. Tanabe, Russell E. Ware, M. Hassan Murad, Jonathan C. Goldsmith, Eduardo Ortiz, Robinson Fulwood, Ann Horton et Joylene John-Sowah, « Management of Sickle Cell Disease. Summary of the 2014 Evidence-Based Report by Expert Panel Members », JAMA, vol. 312, no 10, , p. 1033-1048 (PMID 25203083, DOI 10.1001/jama.2014.10517, lire en ligne)
- (en) Pasquale Niscola, Francesco Sorrentino, Laura Scaramucci, Paolo De Fabritiis et Paolo Cianciulli, « Pain Syndromes in Sickle Cell Disease: An Update », Pain Medicine, vol. 10, no 3, , p. 470-480 (PMID 19416438, DOI 10.1111/j.1526-4637.2009.00601.x, lire en ligne)
- (en) Valentine Brousse, Julie Makani et David C. Rees, « Management of sickle cell disease in the community », BMJ, vol. 348, , article no g1765 (PMID 24613806, DOI 10.1136/bmj.g1765, lire en ligne)
- (en) Joshua J. Field, Paul F. Austin, Ping An, Yan Yan et Michael R. DeBaun, « Enuresis Is a Common and Persistent Problem Among Children and Young Adults with Sickle Cell Anemia », Urology, vol. 72, no 1, , p. 81-84 (PMID 18384865, PMCID 3729268, DOI 10.1016/j.urology.2008.02.006, lire en ligne)
- (en) Russell E. Ware, Renee C. Rees, Sharada A. Sarnaik, Rathi V. Iyer, Ofelia A. Alvarez, James F. Casell, Barry L. Shulkin, Eglal Shalaby-Rana, C. Frederic Strife, John H. Miller, Peter A. Lane, Winfred C. Wang, Scott T. Miller et les investigateurs du BABY HUG, « Renal Function in Infants with Sickle Cell Anemia: Baseline Data from the BABY HUG Trial », The Journal of Pediatrics, vol. 156, no 1, , p. 66-70.e1 (PMID 19880138, PMCID 4755353, DOI 10.1016/j.jpeds.2009.06.060, lire en ligne)
- (en) Ade Olujohungbe et Arthur L. Burnett, « How I manage priapism due to sickle cell disease », British Journal of Haematology, vol. 160, no 6, , p. 754-765 (PMID 23293942, DOI 10.1111/bjh.12199, lire en ligne)
- (en) J. Glassberg, « Evidence-based management of sickle cell disease in the emergency department », Emergency Medicine Practice, vol. 13, no 8, , p. 1-20 (PMID 22164362)
- (en) Scott T. Miller, « How I treat acute chest syndrome in children with sickle cell disease », Blood, vol. 117, no 20, , p. 5297-5305 (PMID 21406723, DOI 10.1182/blood-2010-11-261834, lire en ligne)
- (en) Armand Mekontso Dessap, Rusel Leon, Anoosha Habibi, Ruben Nzouakou, Françoise Roudot-Thoraval, Serge Adnot, Bertrand Godeau, Frederic Galacteros, Christian Brun-Buisson, Laurent Brochard et Bernard Maître, « Pulmonary hypertension and cor pulmonale during severe acute chest syndrome in sickle cell disease », American Journal of Respiratory and Critical Care Medicine, vol. 177, no 6, (PMID 18174543, DOI 10.1164/rccm.200710-1606OC)
- (en) Rabindra N. Paul, Oswaldo L. Castro, Anita Aggarwal et Patricia A. Oneal, « Acute chest syndrome: sickle cell disease », European Journal of Haematology, vol. 87, no 3, , p. 191-207 (PMID 21615795, DOI 10.1111/j.1600-0609.2011.01647.x, lire en ligne)
- (en) Kofi A. Anie et John Green, « Psychological therapies for sickle cell disease and pain », Cochrane Database of Systematic Reviews, vol. 5, , article no CD001916 (PMID 25966336, DOI 10.1002/14651858.CD001916.pub3, lire en ligne)
- (en) Howard A. Pearson, « Sickle Cell Anemia and Severe Infections Due to Encapsulated Bacteria », The Journal of Infectious Diseases, vol. 136, no Supplement 1, , S25-S30 (PMID 330779, DOI 10.1093/infdis/136.Supplement.S25, lire en ligne)
- (en) Wing Yen Wong, Darleen R. Powars, Linda Chan, Alan Hiti, Cage Johnson et Gary Overturf, « Polysaccharide encapsulated bacterial infection in sickle cell anemia: A thirty year epidemiologic experience », American Journal of Hematology, vol. 39, no 3, , p. 176-182 (PMID 1546714, DOI 10.1002/ajh.2830390305, lire en ligne)
- (en) Mariane de Montalembert, « Management of sickle cell disease », BMJ, vol. 337, , article no a1397 (PMID 18779222, DOI 10.1136/bmj.a1397, JSTOR 20510824, lire en ligne)
- (en) Rana Khatib, Raja Rabah et Sharada A. Sarnaik, « The spleen in the sickling disorders: an update », Pediatric Radiology, vol. 39, no 1, , p. 17-22 (PMID 19002450, DOI 10.1007/s00247-008-1049-9, lire en ligne)
- (en) Svetoslav N. Slavov, Simone Kashima, Ana Cristina Silva Pinto et Dimas Tadeu Covas, « Human parvovirus B19: general considerations and impact on patients with sickle-cell disease and thalassemia and on blood transfusions », FEMS Immunology & Medical Microbiology, vol. 62, no 3, , p. 247-262 (PMID 21585562, DOI 10.1111/j.1574-695X.2011.00819.x, lire en ligne)
- (en) R. S. Balgir., « Community Expansion and Gene Geography of Sickle Cell Trait and G6PD Deficiency, and Natural Selection against Malaria: Experience from Tribal Land of India », Cardiovascular & Hematological Agents in Medicinal Chemistry, vol. 10, no 1, , p. 3-13 (PMID 22264009, DOI 10.2174/187152512799201190, lire en ligne)
- (en) T. Jadavji et C. G. Prober, « Dactylitis in a child with sickle cell trait », Canadian Medical Association Journal, vol. 132, no 7, , p. 814-815 (PMID 3978504, PMCID 1345873, lire en ligne)
- (en) V. T. Worrall et V. Butera, « Sickle-cell dactylitis », The Journal of Bones and Joints Surgery. American Volume., vol. 58, no 8, , p. 1161-1163 (PMID 1002763)
- (en) B. Balkaran, G. Char, J. S. Morris, P. W. Thomas, B. E. Serjeant et G. R. Serjeant, « Stroke in a cohort of patients with homozygous sickle cell disease », The Journal of Pediatrics, vol. 120, no 3, , p. 360-366 (PMID 1538280, DOI 10.1016/S0022-3476(05)80897-2, lire en ligne)
- (en) Mark T. Gladwin, Vandana Sachdev, Maria L. Jison, Yukitaka Shizukuda, Jonathan F. Plehn, Karin Minter, Bernice Brown, Wynona A. Coles, James S. Nichols, Inez Ernst, Lori A. Hunter, William C. Blackwelder, Alan N. Schechter, Griffin P. Rodgers, Oswaldo Castro et Frederick P. Ognibene, « Pulmonary Hypertension as a Risk Factor for Death in Patients with Sickle Cell Disease », The New England Journal of Medicine, vol. 350, no 9, , p. 886-895 (PMID 14985486, DOI 10.1056/NEJMoa035477, lire en ligne)
- (en) Florence Parent, Dora Bachir, Jocelyn Inamo, François Lionnet, Françoise Driss, Gylna Loko, Anoosha Habibi, Soumiya Bennani, Laurent Savale, Serge Adnot, Bernard Maitre, Azzedine Yaïci, Leila Hajji, Dermot S. O'Callaghan, Pierre Clerson, Robert Girot, Frederic Galacteros et Gerald Simonneau, « A Hemodynamic Study of Pulmonary Hypertension in Sickle Cell Disease », The New England Journal of Medicine, vol. 365, no 1, , p. 44-53 (PMID 21732836, DOI 10.1056/NEJMoa1005565, lire en ligne)
- Hammoudi N, Lionnet F, Redheuil A, Montalescot, Cardiovascular manifestations of sickle cell disease, Eur Heart J, 2020;41:1365–1373
- (en) Jon I. Scheinman, « Sickle cell disease and the kidney », Nature Clinical Practice. Nephrology, vol. 5, no 2, , p. 78-88 (PMID 19048000, DOI 10.1038/ncpneph1008, lire en ligne)
- (en) Nicolas Leveziel, Sylvie Bastuji-Garin, Franck Lalloum, Giuseppe Querques, Pascale Benlian, Michel Binaghi, Gabriel Coscas, Gisèle Soubrane, Dora Bachir, Frédéric Galactéros et Eric Souied, « Clinical and Laboratory Factors Associated With the Severity of Proliferative Sickle Cell Retinopathy in Patients With Sickle Cell Hemoglobin C (SC) and Homozygous Sickle Cell (SS) Disease », Medicine, vol. 90, no 6, , p. 372-378 (PMID 22033449, DOI 10.1097/MD.0b013e3182364cba, lire en ligne)
- (en) Orah S. Platt, Donald J. Brambilla, Wendell F. Rosse, Paul F. Milner, Oswaldo Castro, Martin H. Steinberg et Panpit P. Klug, « Mortality In Sickle Cell Disease — Life Expectancy and Risk Factors for Early Death », The New England Journal of Medicine, vol. 330, no 23, , p. 1639-1644 (PMID 7993409, DOI 10.1056/NEJM199406093302303, lire en ligne)
- (en) Orah S. Platt, « Hydroxyurea for the Treatment of Sickle Cell Anemia », The New England Journal of Medicine, vol. 358, no 13, , p. 1362-1369 (PMID 18367739, DOI 10.1056/NEJMct0708272, lire en ligne)
- (en) Nancy S. Green, Mary E. Fabry, Lazar Kaptue‐Noche et Ronald L. Nagel, « Senegal haplotype is associated with higher HbF than benin and cameroon haplotypes in African children with sickle cell anemia », American Journal of Hematology, vol. 44, no 2, , p. 145-146 (PMID 7505527, DOI 10.1002/ajh.2830440214, lire en ligne)
- Emmanuelle Lesprit et Philippe Lesprit, « Infections bactériennes dans la drépanocytose : Drépanocytose », La Revue du praticien, vol. 54, no 14, , p. 1574-1577
- (en) Anthony C. Allison, « Genetic control of resistance to human malaria », Current Opinion in Immunology, vol. 21, no 5, , p. 499-505 (PMID 19442502, DOI 10.1016/j.coi.2009.04.001, lire en ligne)
- Pablo Bartolucci, « Hémoglobinopathies : un avantage contre le paludisme mais au risque de maladies sévères », La Revue du praticien, vol. 64, no 8, , p. 1110-1111 (lire en ligne)
- (en) Dominic P. Kwiatkowski, « How Malaria Has Affected the Human Genome and What Human Genetics Can Teach Us about Malaria », American Journal of Human Genetics, vol. 77, no 2, , p. 171-192 (PMID 16001361, DOI 10.1086/432519, Bibcode 1224522, lire en ligne)
- (en) Nicolas Ponçon, Céline Toty, Grégory L'Ambert, Gilbert Le Goff, Cécile Brengues, Francis Schaffner et Didier Fontenille, « Biology and dynamics of potential malaria vectors in Southern France », Malaria Journal, vol. 6, no 18, (PMID 17313664, DOI 10.1186/1475-2875-6-18, Bibcode 1808464, lire en ligne)
- (en) Elliott Vichinsky, Deborah Hurst, Ann Earles, Klara Kleman et Bertram Lubin, « Newborn Screening for Sickle Cell Disease: Effect on Mortality », Pediatrics, vol. 81, no 6, , p. 749-755 (PMID 3368274, lire en ligne)
- (en) E. G. Davies, C. Riddington, R. Lottenberg et N. Dower, « Pneumococcal vaccines for sickle cell disease », The Cochrane Database of Systematic Reviews, vol. 1, , article no CD003885 (PMID 14974042, DOI 10.1002/14651858.CD003885.pub2, lire en ligne)
- Justine Gellen-Dautremer, Valentine Brousse et Jean-Benoît Arlet, « Complications aiguës de la drépanocytose », La Revue du praticien, vol. 64, no 8, , p. 1114-1119
- (en) « Higher Doses Of Morphine Justified For Sickle Cell Patients », sur , (consulté le ).
- (en) Robert J. Adams, Virgil C. McKie, Lewis Hsu, Beatrice Files, Elliott Vichinsky, Charles Pegelow, Miguel Abboud, Dianne Gallagher, Abdullah Kutlar, Fenwick T. Nichols, Duane R. Bonds, Donald Brambilla, Gerald Woods, Nancy Olivieri, Catherine Driscoll, Scott Miller, Winfred Wang, Anne Hurlett, Charles Scher, Brian Berman, Elizabeth Carl, Anne M. Jones, E. Steve Roach, Elizabeth Wright, Robert A. Zimmerman et Myron Waclawiw, « Prevention of a First Stroke by Transfusions in Children with Sickle Cell Anemia and Abnormal Results on Transcranial Doppler Ultrasonography », The New England Journal of Medicine, vol. 339, no 1, , p. 5-11 (PMID 9647873, DOI 10.1056/NEJM199807023390102, lire en ligne)
- (en) L. M. Rabb, Yvonne Grandison, Karlene Mason, R. J. Hayes, Beryl Serjeant et G. R. Serjeant, « A trial of folate supplementation in children with homozygous sickle cell disease », British Journal of Haematology, vol. 54, no 4, , p. 589-594 (PMID 6347243, DOI 10.1111/j.1365-2141.1983.tb02138.x, lire en ligne)
- (en) Oluseyi Oniyangi et Aika A. A. Omari, « Malaria chemoprophylaxis in sickle cell disease », The Cochrane Database of Systematic Reviews, vol. 18, no 4, , article no CD003489 (PMID 17054173, DOI 10.1002/14651858.CD003489.pub2, lire en ligne)
- (en) Arturo J. Martí‐Carvajal, Ivan Solà et Luis H Agreda‐Pérez, « Treatment for avascular necrosis of bone in people with sickle cell disease », The Cochrane Database of Systematic Reviews, no 8, , article no CD004344 (PMID 27502327, DOI 10.1002/14651858.CD004344.pub6, lire en ligne)
- (en) Samuel Charache, Michael L. Terrin, Richard D. Moore, George J. Dover, Franca B. Barton, Susan V. Eckert, Robert P. McMahon, Ph.D., Duane R. Bonds et les investigateurs du Multicenter Study of Hydroxyurea in Sickle Cell Anemia, « Effect of Hydroxyurea on the Frequency of Painful Crises in Sickle Cell Anemia », The New England Journal of Medicine, vol. 332, no 20, , p. 1317-1322 (PMID 7715639, DOI 10.1056/NEJM199505183322001, lire en ligne)
- (en) Martin H. Steinberg, Franca Barton, Oswaldo Castro, Charles H. Pegelow, Samir K. Ballas, Abdullah Kutlar, Eugene Orringer, Rita Bellevue, Nancy Olivieri, James Eckman, Mala Varma, Gloria Ramirez, Brian Adler, Wally Smith, Timothy Carlos, Kenneth Ataga, Laura DeCastro, Carolyn Bigelow, Yogen Saunthararajah, Margaret Telfer, Elliott Vichinsky, Susan Claster, Susan Shurin, Kenneth Bridges, Myron Waclawiw, Duane Bonds et Michael Terrin, « Effect of Hydroxyurea on Mortality and Morbidity in Adult Sickle Cell Anemia. Risks and Benefits Up to 9 Years of Treatment », JAMA, vol. 289, no 13, , p. 1645-1651 (PMID 12672732, DOI 10.1001/jama.289.13.1645, lire en ligne)
- (en) O. S. Platt, S. H. Orkin, G. Dover, G. P. Beardsley, B. Miller et D. G. Nathan, « Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia », The Journal of Clinical Investigation, vol. 74, no 2, , p. 652-656 (PMID 6205021, DOI 10.1172/JCI111464, lire en ligne)
- (en) Richard D. Moore, Samuel Charache, Michael L. Terrin, Franca B. Barton, Samir K. Ballas, E. Orringer, S. Jones, D. Strayhorn, W. Rosse, G. Phillips, D. Peace, A. Johnson-Telfair, P. Milner, A. Kutlar, A. Tracy, S. K. Ballas, G. E. Allen, J. Moshang, B. Scott, M. Steinberg et al., « Cost-Effectiveness of Hydroxyurea in Sickle Cell Anemia », American Journal of Hematology, vol. 64, no 1, , p. 26-31 (PMID 10815784, DOI 10.1002/(SICI)1096-8652(200005)64:1<26::AID-AJH5>3.0.CO;2-F, lire en ligne)
- (en) Frédéric B. Piel, Martin H. Steinberg et David C. Rees, « Sickle Cell Disease », The New England Journal of Medicine, vol. 376, no 16, , p. 1561-1573 (PMID 28423290, DOI 10.1056/NEJMra1510865, lire en ligne)
- (en) « FDA approves new treatment for sickle cell disease », sur www.fda.gov, (consulté le ).
- (en) Yutaka Niihara, Han A. Koh, Lan Tran, Rafael Razon, Henry Macan, Charles Stark, Ted Wun et Patricia Adams-Graves, « A Phase 3 Study of L-Glutamine Therapy for Sickle Cell Anemia and Sickle ß0-Thalassemia », Blood, vol. 124, no 21, , p. 86 (lire en ligne)
- Noir&Blanc Escroquerie : le VK500
- Malika Benkerrou, « Indications des allogreffes de cellules souches hématopoïétiques dans la drépanocytose », La Revue du Praticien, , p. 1130-1131.
- Site des hôpitaux de Lyon, consulté le 30 avril 2008]
- « Tout savoir sur le don | Don de Moelle Osseuse », sur www.dondemoelleosseuse.fr (consulté le )
- (en) Robert Pawliuk, Karen A. Westerman, Mary E. Fabry, Emmanuel Payen, Robert Tighe, Eric E. Bouhassira, Seetharama A. Acharya, James Ellis, Irving M. London, Connie J. Eaves, R. Keith Humphries, Yves Beuzard, Ronald L. Nagel et Philippe Leboulch, « Correction of Sickle Cell Disease in Transgenic Mouse Models by Gene Therapy », Science, vol. 294, no 5550, , p. 2368-2371 (PMID 11743206, DOI 10.1126/science.1065806, Bibcode 2001Sci...294.2368P, lire en ligne)
- (en) Arthur Bank, Ronald Dorazio et Philippe Leboulch, « A Phase I/II Clinical Trial of β-Globin Gene Therapy for β-Thalassemia », Annals of the New York Academy of Science, vol. 1054, no 1, , p. 308-316 (PMID 16339679, DOI 10.1196/annals.1345.007, Bibcode 2005NYASA1054..308B, lire en ligne)
- Sandrine Cabut, « Drépanocytose : succès d’une thérapie génique chez le premier patient traité », sur www.lemonde.fr, (consulté le ).
- (en) Jean-Antoine Ribeil, Salima Hacein-Bey-Abina, Emmanuel Payen, Alessandra Magnani, Michaela Semeraro, Elisa Magrin, Laure Caccavelli, Benedicte Neven, Philippe Bourget, Wassim El Nemer, Pablo Bartolucci, Leslie Weber, Hervé Puy, Jean-François Meritet, David Grevent, Yves Beuzard, Stany Chrétien, Thibaud Lefebvre, Robert W. Ross, Olivier Negre, Gabor Veres, Laura Sandler, Sandeep Soni, Mariane de Montalembert, Stéphane Blanche, Philippe Leboulch et Marina Cavazzana, « Gene Therapy in a Patient with Sickle Cell Disease », The New England Journal of Medicine, vol. 376, no 9, , p. 848-855 (PMID 28249145, DOI 10.1056/NEJMoa1609677, lire en ligne)
- « La drépanocytose : traitements et bonnes pratiques », sur Association Pour l'Information et la Prévention de la Drépanocytose (APIDP), (consulté le ).
- (en) « Sickle-cell anaemia », sur www.who.int, (consulté le ).
- Gil Tchernia, « La longue histoire de la drépanocytose », La Revue du praticien, vol. 54, no 14, , p. 1618-1621
- (en) Mitch Leslie, « 170-year-old mystery solved: Why deer have deformed blood cells just like some people », sur www.sciencemag.org, (DOI 10.1126/science.aar7918, consulté le ).
- (en) Wasil Jastaniah, « Epidemiology of sickle cell disease in Saudi Arabia », Annals of Saudi Medicine, vol. 31, no 3, , p. 289-293 (PMID 21623060, PMCID 3119971)
- (en) Ziad Ahmed Memish et Mohammad Y. Saeedi, « Six-year outcome of the national premarital screening and genetic counseling program for sickle cell disease and β-thalassemia in Saudi Arabia », Annals of Saudi Medicine, vol. 31, no 3, , p. 229-235 (PMID 21623050, PMCID 3119961)
- (en) Sheikha Salim Al Arrayed1 et Neva Haites, « Features of sickle-cell disease in Bahrain », La Revue de Santé de la Méditerranée orientale, vol. 1, no 1, , p. 112-119 (lire en ligne)
- (en) Shaikha Al Arrayed et Amani Al Hajeri, « Public awareness of sickle cell disease in Bahrain », Annals of Saudi Medicine, vol. 30, no 4, , p. 284-288 (PMID 20622345, PMCID 2931779)
- (en) Neeraj Awasthy, K. C. Aggarwal, P. C. Goyal, M. S. Prasad, S. Saluja et M. Sharma, « Sickle cell disease: Experience of a tertiary care center in a nonendemic area », Annals of Tropical Medicine and Public Health, vol. 1, no 1, , p. 1-4 (DOI 10.4103/1755-6783.43069, lire en ligne)
- (en) Frédéric B. Piel, Andrew J. Tatem, Zhuojie Huang, Sunetra Gupta, Thomas N. Williams et David J. Weatherall, « Global migration and the changing distribution of sickle haemoglobin: a quantitative study of temporal trends between 1960 and 2000 », The Lancet Global Health, vol. 2, no 2, , e80-e89 (PMID 24748392, DOI 10.1016/S2214-109X(13)70150-5, Bibcode 3986033, lire en ligne)
- (en) « Sickle Cell Disease (SCD) Data & Statistics », sur www.cdc.gov, (consulté le ).
- (en) Salzano Francisco, « Incidence, effects, and management of sickle cell disease in Brazil », Journal of Pediatric Hematology/Oncology, vol. 7, no 3, , p. 240-244 (PMID 2416230, lire en ligne)
- Bardakdjian, Josiane et Wajcman, Henri « Épidémiologie de la drépanocytose » La Revue du Praticien 2004
- (de) R. Dickerhoff et A. von Ruecker A., « Manifestations of sickle cell disease in adolescents and young adults. Clinical aspects and therapy references », Klinische Padiatrie, vol. 210, no 1, , p. 10-16 (PMID 9522298)
- (en) Joachim B. Kunz, Holger Cario, Regine Grosse, Andrea Jarisch, Stephan Lobitz et Andreas E. Kulozik, « The epidemiology of sickle cell disease in Germany following recent large-scale immigration », Pediatric Blood & Cancer, vol. 64, no 7, , article no e26550 (PMID 28383793, DOI 10.1002/pbc.26550, lire en ligne)
- (en) « Screening for sickle cell and thalassaemia – Prevalence », sur Public Health England (consulté le ).
- (en) D. Lena-Russo, M. L. North et R. Girot, « Épidémiologie des maladies génétiques de l'hémoglobine en France métropolitaine », La Revue du praticien, vol. 42, no 15, , p. 1867-1872
- « Dépistage néonatal de la drépanocytose en France — Pertinence d’une généralisation du dépistage à l’ensemble des nouveau-nés » [PDF], sur www.has-sante.fr, (consulté le ).
- « AFDPHE, Bilan d'Activité 2016 », sur , Association Française pour le Dépistage et les Prévention des Handicaps de l'Enfant, (consulté le ).
- (en) National Institutes of Health, Consensus, « Consensus conference. Newborn screening for sickle cell disease and other hemoglobinopathies », Journal of American Medical Association, vol. 258, no 9, , p. 1205-1209 (PMID 3626004)
- L. S. Zohoun, G. Mérault, P. Reinette et J. Rosa, « Politiques de santé et drépanocytose », La Revue du praticien, vol. 42, no 15, , p. 1873-1877
- Josiane Bardakdjian-Michau, « Le dépistage néonatal de la drépanocytose en France », Archives de Pédiatrie, vol. 15, no 5, , p. 738-740 (DOI 10.1016/S0929-693X(08)71894-3, lire en ligne)
- « Drépanocytose : un test de dépistage rapide à l'essai sur le continent », JeuneAfrique.com, (lire en ligne, consulté le )
- (en) Nathaniel Z. Piety, Xiaoxi Yang, Julie Kanter, Seth M. Vignes, Alex George et Sergey S. Shevkoplyas, « Validation of a Low-Cost Paper-Based Screening Test for Sickle Cell Anemia », PLoS One, vol. 11, no 1, , article no e0144901 (PMID 26735691, PMCID 4703210, DOI 10.1371/journal.pone.0144901, Bibcode 2016PLoSO..1144901P, lire en ligne)
- (en) Robert Lebby, « Case of absence of the spleen », The Southern Journal of Medicine and Pharmacy, vol. 1, , p. 481-483
- (en) Samir K. Ballas, Kalpna Gupta et Patricia Adams-Graves, « Sickle cell pain: a critical reappraisal », Bood, vol. 120, no 18, , p. 3647-3656 (PMID 22923496, DOI 10.1182/blood-2012-04-383430, lire en ligne)
- A. Lainé, « Entretien : " Il n'y a pas de maladie plus racialisée que la drépanocytose ". », La Revue du Praticien - Médecine Générale. « 26 », no 876, , p.138-139
- (en) Todd L. Savitt et Morton F. Goldberg, « Herrick's 1910 Case Report of Sickle Cell Anemia. The Rest of the Story », JAMA, vol. 261, no 2, , p. 266-271 (PMID 2642320, DOI 10.1001/jama.1989.03420020120042, lire en ligne)
- (en) James B. Herrick, « Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia », JAMA, vol. 74, no 3, , p. 179-184 (PMID 11501714, PMCID 2588723, DOI 10.1001/archinte.1910.00050330050003, lire en ligne)
- (en) Graham R. Serjeant, « One hundred years of sickle cell disease », British Journal of Haematology, vol. 151, no 5, , p. 425-429 (PMID 20955412, DOI 10.1111/j.1365-2141.2010.08419.x, lire en ligne)
- (en) James B. Herrick, « Peculiar Elongated and Sickle-Shaped Red Blood Corpuscles in a Case of Severe Anemia », JAMA, vol. 312, no 10, , p. 1063 (PMID 25203098, DOI 10.1001/jama.2014.11011, lire en ligne)
- (en) R. E. Washburn, « Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia », Virginia Medical Semi-Monthly, vol. 15, , p. 490-493
- (en) Verne Rheem Mason, « Landmark article Oct. 14, 1922: Sickle cell anemia. By V.R. Mason », JAMA, vol. 254, no 14, , p. 1955-1957 (PMID 3900438, DOI 10.1001/jama.1922.02640160038012, lire en ligne)
- D. Labie, « Histoire génétique de la drépanocytose », La Revue du praticien, vol. 42, no 15, , p. 1879-1884
- (en) James V. Neel, « The Inheritance of Sickle Cell Anemia », Science, vol. 110, no 2846, , p. 64-66 (PMID 17774955, DOI 10.1126/science.110.2846.64, JSTOR 1677755, Bibcode 1949Sci...110...64N, lire en ligne)
- (en) Linus Pauling, Harvey Akio Itano, Seymour Jonathan Singer et Ibert C. Wells, « Sickle Cell Anemia, a Molecular Disease », Science, vol. 110, no 2865, , p. 543-548 (PMID 15395398, DOI 10.1126/science.110.2865.543, lire en ligne)
- (en) Vernon M. Ingram, « A Specific Chemical Difference Between the Globins of Normal Human and Sickle-Cell Anæmia Hæmoglobin », Nature, vol. 178, no 4537, , p. 792-794 (PMID 13369537, DOI 10.1038/178792a0, lire en ligne)
- Site de la Fondation Louis Jeantet
- J. Bernard, Le sang et l'histoire, Buchet / Chastel, , p. 81-91
- (en) Keith Wailoo, « Sickle Cell Disease - A History of Progress and Peril », The New England Journal of Medicine, vol. 376, no 9, , p. 805-807.
- F. Galactéros, « Drépanocytose », La Revue du Praticien, vol. 54, no 14, , p. 1529-1530
- « Prodigy: The Truth About Sickle Cell », interview par Allhiphop.com, 4 février 2008.
- « Tsonga évoque la drépanocytose, la maladie qui le met HS quand il prend l'avion » sur 20minutes.fr, 14 janvier 2019.
- (en) « The Life of Bolaji Badejo », sur Strange Shapes, (consulté le )
Voir aussi
Liens externes
- (en) NCBI (NIH): Bibliographie sur Medline (en anglais) ;
- (en) Rapport de l'OMS (2006).
- Ressources françaises :
- (fr) Contacts en Afrique et en France sur la page de l'association DREPAVIE
- (fr) Orphanet: Informations sur la drépanocytose (consultations, recherches, associations…) ;
- (fr) APIPD – Association pour l'information et la prévention de la drépanocytose ;
- (fr) CIDD – Centre d'information et de dépistage de la drépanocytose ;
- (fr) Afrik.com – dossier complet sur la drépanocytose ;
- (fr) RoFSED – Réseau ouest-francilien de soin des enfants drépanocytaires.
Bibliographie
- K. Wailoo, Dying in the city of the blues : sickle cell anemia and the politics of race and death, The University of North Carolina Press, 2001.
- A. Lainé. La drépanocytose, regards croisés sur une maladie orpheline, Karthala, 2004.
 Portail de la médecine
Portail de la médecine  Portail de l’hématologie
Portail de l’hématologie