Cholangite sclérosante primitive
La cholangite sclérosante primitive (CSP) est une maladie caractérisée par une atteinte inflammatoire idiopathique et fibrosante des voies biliaires intra et/ou extrahépatiques. L’évolution de cette affection cholestatique chronique est variable mais peut se faire vers la cirrhose et les complications graves des hépatopathies. De nombreux aspects de cette maladie restent inconnus ou imprécis.
Pour les articles homonymes, voir CSP.
| Spécialité | Gastro-entérologie |
|---|
| CISP-2 | D98 |
|---|---|
| CIM-10 | K83.0 |
| CIM-9 | 576.1 |
| OMIM | et 613806 602114 et 613806 |
| DiseasesDB | 10643 |
| MedlinePlus | 000285 |
| eMedicine | 187724 |
| eMedicine | med/3556 |
| MeSH | D015209 |
| Patient UK | Primary-sclerosing-cholangitis-pro |
![]() Mise en garde médicale
Mise en garde médicale
Éléments épidémiologiques
La CSP est une maladie rare du sujet jeune (âge moyen de 40 ans au moment du diagnostic[1]) touchant plutôt l'homme (2/3 des cas[1]). Contrairement à la cirrhose biliaire primitive (CBP), cette maladie peut atteindre l’enfant. Tous les types ethniques peuvent être touchés. L'une de ses caractéristiques est son association fréquente (2/3 des cas en France) à une maladie inflammatoire de l'intestin (Rectocolite Hémorragique principalement). Son incidence est estimée autour de 1/100 000 aux États-Unis[2] et en Europe, avec une tendance à l'augmentation[1]. Quoi qu’il en soit, ces données font considérer la CSP comme une maladie orpheline environ trois fois moins fréquente que la CBP.
Elle est souvent associée avec une maladie inflammatoire chronique intestinale[3].
Physiopathologie
La maladie entraîne une lésion des voies biliaires de type inflammatoire, entraînant un rétrécissement de ces dernières, voire leur occlusion. Cela a pour conséquence une augmentation de la pression, avec parfois rupture des canaux biliaires, atteinte hépatique et formation d'une fibrose de l'organe conduisant à une cirrhose. L'évolution peut se faire également vers un cholangiocarcinome (cancer des voies biliaires).
La pathogénie de la CSP n’est pas connue bien que des mécanismes immunologiques et non immunologiques aient été suggérés.
Diverses hypothèses ont été émises dont les principales sont les suivantes : bactériémie portale chronique, métabolisme anormal des acides biliaires par la flore intestinale, production de toxines par cette flore, composition anormale de la bile, infections virales chroniques méconnues, ischémie des voies biliaires, association avec d'autres maladies autoimmunes comme la rectocolite hémorragique, la maladie de Crohn, un syndrome de Gougerot-Sjögren, cas rare d'overlap syndrome avec une hépatite autoimmune, et enfin anomalies génétiques de l’immunorégulation… Très schématiquement, on considère qu’il pourrait s’agir d’une réponse inflammatoire inadaptée à des agents d’origine intestinale chez des patients ayant une susceptibilité génétique particulière. Comme dans d’autres maladies autoimmunes, l’association à certains groupes HLA B a été bien documentée[4]. La mutation de certains gènes semblent favoriser la maladie : c'est le cas sur les gènes IL2, REL, CARD9[5], GPR35, TCF4[6], MMEL1, TNFRSF14, FUT2[7] ainsi que sur d'autres loci[8].
Diagnostic de la CSP
Le mode de révélation est extrêmement variable. Schématiquement le diagnostic est évoqué dans trois grandes circonstances :
- anomalies des tests hépatiques chez des malades asymptomatiques ayant une colite inflammatoire ;
- symptomatologie biliaire (angiocholite ou ictère franc, prurit isolé plus rarement que dans la CBP) ;
- tableau d'hépatopathie chronique non spécifique (hépatite chronique, cirrhose éventuellement compliquée). En outre, des formes particulières ont été individualisées (cf. infra).
Le diagnostic repose classiquement sur l'association de quatre types de signes :
- biologiques (cholestase) ;
- radiologiques (anomalies des voies biliaires intra- ou extrahépatiques) ;
- histologiques (cholangite fibreuse et oblitérante) ;
- association à une autre maladie, et en particulier à une maladie inflammatoire du côlon. L’ensemble de ces quatre signes n’est observé que dans les formes caricaturales et on considère maintenant que le diagnostic de CSP peut être retenu en présence de deux (incluant au moins un critère histologique ou radiologique) de ces quatre critères, en l'absence d'autre étiologie identifiable.
La corrélation entre les signes biologiques, histologiques et radiologiques est faible.
Biologie
Contrairement à la cirrhose biliaire primitive (CBP), il n’existe pas d’anticorps anti-tissus caractéristiques et quasi constants. En effet, la sensibilité des anti-cytoplasmes des polynucléaires neutrophiles de type périnucléaire (pANCA) est très variable selon les séries (26 à 85 %) et leur spécificité est médiocre car ils sont observés également dans les colites inflammatoires et les hépatites auto-immunes.
Anatomopathologie
Les lésions histologiques élémentaires sont au nombre de quatre : la fibrose péricanalaire avec cholangite ou atrophie des cellules biliaires, la prolifération néoductulaire, la diminution du nombre des canaux biliaires et la nécrose hépatocytaire parcellaire en bordure de l’espace porte. Ces lésions sont diversement associées. Une classification en 4 stades, proche de celle de la CBP, a été proposée, le stade IV correspondant à la cirrhose. La lésion la plus évocatrice, la cholangite fibreuse et oblitérante est absente sur la ponction biopsie hépatique dans plus de 2/3 des cas du fait de la répartition hétérogène des lésions à l'intérieur du foie et 5 à 10 % des biopsies sont normales. En conséquence, une biopsie hépatique non évocatrice voire normale ne doit pas faire éliminer le diagnostic de CSP.
Imagerie
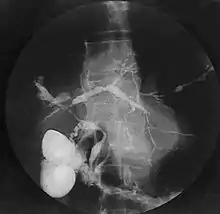
La mise en évidence d’anomalies des voies biliaires reste un élément clé du diagnostic malgré l’existence de formes particulières. L’examen classique de référence est l’opacification des voies biliaires, le plus souvent par cathétérisme rétrograde. Cet examen est techniquement difficile et a une morbidité certaine notamment en termes d’angiocholite (infection des voies biliaires). Il est désormais possible de visualiser les voies biliaires par un examen non invasif, la cholangio IRM (ou bili IRM) qui a une bonne sensibilité et spécificité[9]. La cholangio IRM tend donc à devenir l’examen de premier intention, le cathétérisme rétrograde n’étant pratiqué qu’en cas de difficulté diagnostique ou à visée thérapeutique. Les anomalies observées sont des sténoses souvent multiples, typiquement sans dilatation d’amont nette ; un aspect en chapelet est très évocateur ; des irrégularités murales, voire des aspects diverticulaires sont possibles. L’atteinte est le plus souvent intra- et extrahépatique, rarement uniquement intrahépatique (< 20 %) ou uniquement extrahépatique (< 10 %). Des atteintes du canal cystique et du canal pancréatique ont été rapportées.
Une colite inflammatoire (habituellement RCH, plus rarement maladie de Crohn ou colite inclassée) est présente dans 2/3 à 3/4 des CSP. L'absence d'association cliniquement patente à une colite inflammatoire doit faire réaliser systématiquement une coloscopie avec biopsies, car la colite est souvent peu active, voire totalement latente bien qu’il s’agisse d’une pancolite de façon quasi constante. Chez les patients ayant une RCH, on estime que la CSP est responsable de 40 % des anomalies chroniques des tests hépatiques, les autres causes étant les suivantes : stéatose, consommation excessive d’alcool, hépatite virale, granulomatose, processus septique ou encore toxicité médicamenteuse. D’autres maladies peuvent beaucoup plus rarement être associées à la CSP : fibrosclérose multifocale, pancréatite, diabète et diverses maladies dysimmunitaires : lupus, polyarthrite rhumatoïde…
Le bilan initial d’une CSP comporte donc habituellement un examen clinique, des examens biologiques (tests hépatiques), des examens d’imagerie (échographie et cholangio-IRM), une ponction biopsie hépatique et une coloscopie (en l’absence de maladie colique connue). La ponction biopsie hépatique n’est pas toujours indispensable mais est recommandée car elle fournit des éléments pronostiques et des arguments diagnostiques dans les formes atypiques.
Aspects particuliers - CSP des petits canaux biliaires
Une image histologique de cholangite sclérosante peut être observée en l'absence d'anomalie radiologique des voies biliaires. Le diagnostic de CSP des petits canaux biliaires (« small-duct primary sclerosing cholangitis »), est classiquement retenu si les critères suivants sont réunis : cholestase biologique, histologie compatible, cholangiographie normale, association à une colite inflammatoire et absence d’autres causes de cholestase. Cependant, les séries récentes ne retiennent plus comme nécessaire l’association à une colite inflammatoire. Cette forme fait discuter les autres causes de cholestase à voies biliaires macroscopiquement normales et en particulier la cirrhose biliaire primitive, la sarcoïdose et les cholangites médicamenteuses. La prévalence est habituellement inférieure à 10 % dans les séries de CSP. Cette forme semble plutôt correspondre à une maladie des voies biliaires ayant une histoire naturelle différente car la survenue d’une atteinte des gros canaux biliaires n’a été rapporté que dans environ 15 % des cas après un suivi moyen d’une dizaine d’années. En outre la survenue d’une hépatopathie évoluée est rare et celle d’un cancer des voies biliaires exceptionnelle.
Forme mixte CSP – hépatite auto-immune
Enfin, il existe des formes mixtes CSP/hépatite auto-immune (« Overlap ») dont le diagnostic est suspecté sur les critères biologiques, immunologiques et histologiques habituels d’hépatite auto-immune (HAI) :
- élévation de l'activité des transaminases supérieure à 5 N ;
- élévation des IgG supérieure à 2 N ou présence d'anti-muscles lisses de spécificité anti-actine ;
- lésions inflammatoires périportales et lobulaires marquées. La prévalence exacte n'est pas connue mais cette forme mixte semble plus souvent observée chez l'enfant et l'adulte jeune. Les corticoïdes ont probablement un effet bénéfique mais l’histoire naturelle et le traitement optimal ne sont pas connus.
Diagnostic différentiel
Il se discute différemment selon la présentation. Devant des anomalies cholangiographiques, les principaux diagnostics différentiels sont :
- un remplissage incomplet de l’arbre biliaire en raison de difficultés techniques ;
- une cholangite aiguë (résolutive) ;
- un cholangiocarcinome, dont le diagnostic différentiel est particulièrement difficile, voire impossible sauf en cas de masse tumorale, et qui peut être associé à une CSP (cf. infra) ;
- une cirrhose (ou une pathologie infiltrative du foie) en cas d’anomalies uniquement intrahépatiques.
Lorsque le diagnostic de cholangite sclérosante est posé, le caractère primitif ou secondaire (lithiase de la voie biliaire principale, antécédents de chirurgie biliaire, injection de produit caustique dans les voies biliaires, infection VIH ou atteinte ischémique dans le cadre d’une chimiothérapie par l’artère hépatique, d’une affection thrombosante type hémoglobinurie paroxystique nocturne, d’une thrombose de l’artère hépatique ou encore de lésions de conservation du greffon) doit être discuté en l’absence de maladie inflammatoire du côlon.
La CSP des petites voies biliaires fait discuter les autres causes de cholestase à voies biliaires macroscopiquement normales et en particulier la CBP, la sarcoïdose et les cholangites médicamenteuses.
Évolution
L'évolution de la CSP se fait habituellement vers l'aggravation, la médiane de survie était classiquement, sans traitement, de 9-12 ans après l’affirmation du diagnostic mais atteint désormais 18 ans dans les séries récentes. Différents modèles pronostiques ont été proposés. Dans l'étude comportant le plus grand nombre de malades, les facteurs pronostiques identifiés étaient l'âge, la bilirubinémie, le stade histologique et la présence d'une splénomégalie (augmentation de la taille de la rate). Une modification de ce score n’incluant plus les données histologiques a été proposé (âge, bilirubinémie, albuminémie, activité des transaminases, hémorragie digestive). Il existe toutefois une grande variabilité individuelle et les modèles pronostiques sont peu utilisés. La CSP et la maladie colique évoluent chacune pour leur propre compte. Cependant, pour les RCH, l'association à une CSP est un facteur de risque particulier de survenue d'un adénocarcinome du côlon[10]. Sans atteinte colique, la CSP ne semble pas augmenter le risque de cancer du côlon[11].
Un fait majeur est la survenue possible d'un cancer des voies biliaires ou cholangiocarcinome. Les grandes séries médicales suggèrent que l’incidence annuelle du cholangiocarcinome est faible, de l'ordre de 1 %[4]. Le diagnostic en est extrêmement difficile en raison des anomalies préexistantes des voies biliaires. Une augmentation de la concentration sanguine de l’ACE et du CA 19.9 peut fournir des éléments d’orientation mais ces marqueurs manquent de sensibilité et de spécificité[12]. Aucun facteur prédictif de survenue n'a été clairement mis en évidence. Le cholangiocarcinome n’est toutefois pas une évolution obligatoire, même dans les CSP évoluées. 30 à 50 % des cholangiocarcinomes sont diagnostiqués dans les deux ans suivant la découverte de la CSP.
En pratique, bien qu’aucune stratégie de surveillance n’ait été validée, l’attitude suivante peut être proposée pour le suivi des CSP :
- tous les 6 mois : examen clinique et tests hépatiques simples (bilirubine, enzymes, électrophorèse des protides, plaquettes, TP) et dosage de l’ACE et du CA19-9 ;
- tous les ans : imagerie des voies biliaires (échographie « experte » ou bili-IRM) ;
- En cas d’association à une maladie inflammatoire du côlon : coloscopie avec biopsies étagées multiples tous les 3 ans à partir de la 10e année d’évolution et tous les 2 ans à partir de la 20e année (voire à une fréquence plus élevée).
Si une majoration des anomalies biologiques est constatée, ou bien sûr en cas d’évènement clinique, il faut s’efforcer de répondre, par les examens appropriés, aux questions suivantes :
- existe-t-il des arguments en faveur d’un cholangiocarcinome ?
- existe-t-il une sténose dominante ou une lithiase biliaire pouvant éventuellement bénéficier d’un traitement mécanique ?
- existe-t-il des arguments en faveur d’une hépatite autoimmune ou d’une hépatotoxicité médicamenteuse (en particulier du traitement de la colite inflammatoire) ?
- quelle est l’observance du traitement ?
Traitement
L'European Association for the Study of the Liver a publié des recommandations pour la prise en charge de la CSP, datant de 2009[13] ainsi que l'American Association for the Study of Liver Diseases, datant de 2010[14].
Traitement médicamenteux
La méconnaissance des causes de la CSP est un obstacle important à l’élaboration de propositions thérapeutiques rationnelles. En outre, l’évaluation des traitements est gênée par l’hétérogénéité et la relative rareté de la maladie. De ce fait, la plupart des essais thérapeutiques n’ont inclus qu’un petit nombre de malades, souvent graves et suivis sur des périodes assez courtes. Différents traitements immunosuppresseurs ou à visée antifibrosante, incluant notamment la D-penicillamine, les corticoïdes (dont le budésonide[15]), la ciclosporine, le méthotrexate et la colchicine ont été testés dans des études ouvertes ou randomisées sans qu’aucun d’entre eux ne fasse la preuve de son efficacité.
L’acide ursodésoxycholique (AUDC) est la principale proposition thérapeutique. En raison de la similitude avec la cirrhose biliaire primitive, l’AUDC a été testé à la même posologie (13-15 mg/kg/j) chez les patients ayant une CSP. L’étude randomisée la plus importante (105 patients, AUDC versus placebo) a confirmé l’effet sur la biologie (diminution de la bilirubinémie, de l’activité des phosphatases alcalines et des transaminases, augmentation de l’albuminémie) mais n’a pas montré de bénéfice en termes de survie sans transplantation[16]. Toutefois, les patients inclus dans cette étude étaient à un stade avancé de la maladie comme en atteste une survie à quatre ans sans transplantation inférieure à 55 % dans le groupe placebo. À plus fortes doses, l'AUDC ne parvient pas à prouver une amélioration de la survie ou un risque moindre d'évolution vers le cholangiocarcinome[17]. Il existe même un doute sur un effet délétère du traitement à hautes doses[18], dont celui d'une majoration du risque de survenue d'un cancer du côlon[19]. En pratique clinique, la quasi-totalité des CSP reçoit actuellement de l’AUDC à des doses modérées en raison notamment de sa très bonne tolérance. Un autre argument en faveur de l’utilisation de l’AUDC est extrahépatique. En effet, deux études ont suggéré que la prise d’AUDC au long cours était associée à une diminution de la prévalence de la dysplasie colique chez les patients ayant une rectocolite hémorragique associée à la CSP. L'utilisation de dérivés de l'AUDC est en cours de test[4].
L'administration d'un antibiotique, le métronidazole à l'AUDC semble améliorer les paramètres hépatiques mais pas la progression de la maladie[20]. D'autres antibiotiques ont été testés avec des résultats comparables[4].
Traitement associé à l’acide ursodésoxycholique
Un traitement associé à l’AUDC peut être proposé dans deux situations :
- sténose unique ou nettement prédominante au niveau du hile ou de la voie biliaire principale : dilatation au ballonnet et/ou prothèse biliaire temporaire[21] ;
- argument en faveur d’une hépatite autoimmune associée : corticoïdes azathioprine.
Le traitement chirurgical se résume désormais pratiquement à la transplantation hépatique qui est la seule option thérapeutique pour les patients ayant atteint le stade terminal de leur hépatopathie. Le bénéfice de la transplantation a été démontré par comparaison de la survie observée chez les malades transplantés à celle prédite par un modèle pronostique en l’absence de transplantation chez ces mêmes malades. Les indications reconnues de la transplantation sont :
- un ictère prolongé avec bilirubinémie > 100 µmol/l ;
- des épisodes répétés d’angiocholites mal contrôlées par les antibiotiques ;
- une cirrhose constituée avec hypertension portale.
Pour certains, le risque de survenue d’un cholangiocarcinome est un argument pour une indication précoce de transplantation mais les critères et le bénéfice d’un tel type d’indication restent à préciser. Le taux de survie à 5 ans des CSP transplantées est supérieur à 70-80 % dans les séries récentes. Le principal facteur pronostique péjoratif est un cholangiocarcinome connu (éventuellement de diagnostic per-opératoire), mais un cholangiocarcinome diagnostiqué uniquement lors de l’examen anatomopathologique de la pièce d’hépatectomie n’est pas associé à une diminution de la survie. Il existe un risque de récidive de la CSP sur le greffon, de l’ordre de 20 % à 5 ans[4], mais cette récidive n’est pas une cause importante de décès ou de retransplantation. Chez les patients transplantés ayant une RCH, une poussée de la maladie intestinale peut être observée malgré le traitement immunosuppresseur et une surveillance coloscopique au moins annuelle est nécessaire en raison d’un risque majoré de cancer du côlon.
Angiocholite et fièvre
Des épisodes fébriles ne sont pas toujours associés à une infection bactérienne et peuvent disparaître spontanément. Les infections bactériennes surviennent essentiellement en cas d'intervention sur les voies biliaires (endoscopique, radiologique ou chirurgicale) ou de présence de calculs dans la voie biliaire principale ou dans les voies biliaires intrahépatiques. Le traitement est antibiotique et, éventuellement, endoscopique interventionnel si des calculs sont présents dans la voie biliaire principale. Il est recommandé aux patients ayant déjà fait un épisode d'angiocholite de se munir d'antibiotiques (type quinolone) en cas de déplacement dans des zones faiblement médicalisées.
Traitement des symptômes et des complications de la cholestase chronique
Le traitement du prurit et de l'éventuel déficit en vitamines liposolubles n'a pas fait l'objet d'étude spécifique à la CSP. Les règles générales sont préconisées : en première ligne, cholestyramine (4 à 16 g/j) prise à distance (2 à 4 heures) de l'AUDC ; en seconde ligne, rifampicine à la posologie de 300 mg/j.
La maladie osseuse associée à la CSP a été peu étudiée. Dans le principal travail, les facteurs associés à l'ostéopénie étaient l'âge, la sévérité de l'hépatopathie et l'ancienneté de la colite inflammatoire. Bien que non identifiées dans cette étude, la ménopause et la corticothérapie sont aussi de très probables facteurs de risque. Malgré l'absence d'évaluation disponible, les recommandations faites dans la CBP peuvent être appliquées : ostéodensitométrie initiale puis tous les 2 à 4 ans, exercice et arrêt du tabac, apport de vitamine D et de calcium, hormonothérapie substitutive en l'absence de contre-indication et enfin, en cas d'ostéoporose démontrée, traitement par biphosphonates.
Notes et références
- (en) Molodecky NA, Kareemi H, Parab R et al. « Incidence of primary sclerosing cholangitis: a systematic review and meta-analysis » Hepatology, 2011;53:1590-1599
- (en) LaRusso NF, Shneider BL, Black D, Gores GJ, James SP, Doo E et al. « Primary sclerosing cholangitis: summary of a workshop » Hepatology, 2006;44:746-64
- (en) Boonstra K, van Erpecum KJ, van Nieuwkerk KM et al. « Primary sclerosing cholangitis is associated with a distinct phenotype of inflammatory bowel disease » Inflamm Bowel Dis, 2012;18:2270-2276
- (en) Hirschfield GM, Karlsen TH, Lindor KD, Adams DH, « Primary sclerosing cholangitis » Lancet, 2013;382:1587-1599
- (en) Janse M, Lamberts LE, Franke L et al. « Three ulcerative colitis susceptibility loci are associated with primary sclerosing cholangitis and indicate a role for IL2, REL, and CARD9 » Hepatology, 2011;53:1977-1985
- (en) Ellinghaus D, Folseraas T, Holm K et al. « Genome-wide association analysis in sclerosing cholangitis and ulcerative colitis identifies risk loci at GPR35 and TCF4 » Hepatology, 2012;10.1002/hep.25977
- (en) Folseraas T, Melum E, Rausch P et al. « Extended analysis of a genome-wide association study in primary sclerosing cholangitis detects multiple novel risk loci » J Hepatol, 2012;57:366-375
- (en) Melum E, Franke A, Schramm C et al. « Genome-wide association analysis in primary sclerosing cholangitis identifies two non-HLA susceptibility loci » Nat Genet, 2011;43:17-19
- (en) Dave M, Elmunzer BJ, Dwamena BA, Higgins PD, « Primary sclerosing cholangitis: meta-analysis of diagnostic performance of MR cholangiopancreatography » Radiology, 2010;256:387-396
- (en) Soetikno RM, Lin OS, Heidenreich PA, Young HS, Blackstone MO, « Increased risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis: a meta-analysis » Gastrointest Endosc, 2002;56:48-54
- (en) Claessen MM, Vleggaar FP, Tytgat KM, Siersema PD, van Buuren HR, « High lifetime risk of cancer in primary sclerosing cholangitis » J Hepatol, 2009;50:158-164
- (en) Sinakos E, Saenger AK, Keach J, Kim WR, Lindor KD, « Many patients with primary sclerosing cholangitis and increased serum levels of carbohydrate antigen 19-9 do not have cholangiocarcinoma » Clin Gastroenterol Hepatol, 2011;9:434-439
- (en) European Association for the Study of the Liver, « EASL Clinical Practice Guidelines: management of cholestatic liver diseases » J Hepatol, 2009;51:237-267
- (en) Chapman R, Fevery J, Kalloo A et al. « Diagnosis and management of primary sclerosing cholangitis » Hepatology, 2010;51:660-678
- (en) van Hoogstraten HJ, Vleggaar FP, Boland GJ et al. « Budesonide or prednisone in combination with ursodeoxycholic acid in primary sclerosing cholangitis: a randomized double-blind pilot study » Am J Gastroenterol, 2000;95:2015-2022
- (en) Lindor KD, Mayo Primary Sclerosing Cholangitis-Ursodeoxycholic Acid Study Group, « Ursodiol for primary sclerosing cholangitis » N Engl J Med, 1997;336:691-695
- (en) Olsson R, Boberg KM, de Muckadell OS et al. « High-dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5-year multicenter, randomized, controlled study » Gastroenterology, 2005;129:1464-1472
- (en) Lindor KD, Kowdley KV, Luketic VA, et al. « High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis » Hepatology, 2009;50:808-814
- (en) Eaton JE, Silveira MG, Pardi DS et al. « High-dose ursodeoxycholic acid is associated with the development of colorectal neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis » Am J Gastroenterol, 2011;106:1638-1645
- (en) Farkkila M, Karvonen AL, Nurmi H et al. « Metronidazole and ursodeoxycholic acid for primary sclerosing cholangitis: a randomized placebo-controlled trial » Hepatology, 2004;40:1379-1386
- (en) Stiehl A, Rudolph G, Kloters-Plachky P, Sauer P, Walker S, « Development of dominant bile duct stenoses in patients with primary sclerosing cholangitis treated with ursodeoxycholic acid: outcome after endoscopic treatment » J Hepatol, 2002;36:151-156
Liens externes
- FILOIE Filière de santé dédiée aux maladies rares du foie
- Association ALBI Association pour la lutte contre les maladies inflammatoires du foie et des voies biliaires
- AFEF Société française d'hépatologie
 Portail de la médecine
Portail de la médecine