Maladie de Kawasaki
La maladie de Kawasaki, ou « syndrome lympho-cutanéo-muqueux » ou « syndrome adéno-cutanéo-muqueux », est une maladie infantile, d'origine immunologique, consistant en une vascularite fébrile touchant les artères de moyen et petit calibre.
| Spécialité | Immunologie |
|---|---|
| Symptôme | Langue framboisée (en) |
| CIM-10 | M30.3 |
|---|---|
| CIM-9 | 446.1 |
| OMIM | 611775 |
| DiseasesDB | 7121 |
| MedlinePlus | 000989 |
| eMedicine | 965367 |
| eMedicine | ped/1236 |
| MeSH | D009080 |
![]()
L'hypothèse dominante est qu'il s'agit d'une maladie infectieuse, peut-être secondaire à une co-infection par plusieurs germes, se traduisant par une inflammation des artères. C'est une maladie émergente en Europe.
Historique
Ce syndrome a été décrit en 1967 au Japon[1] où la maladie est fréquente.
Symptômes et clinique
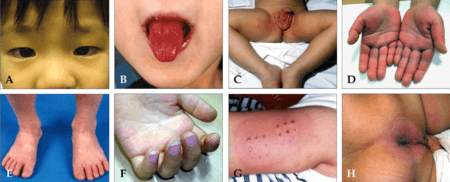
La maladie se caractérise par :
- une forte fièvre (supérieure à 38,5 °C), de plus de cinq jours et ne cédant pas aux médicaments antipyrétiques ou aux antibiotiques. Elle peut être accompagnée d'une altération de l'état général, de maux de tête ;
- une conjonctivite bilatérale bulbaire, dite non-exsudative ;
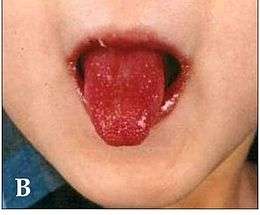
- une inflammation de la muqueuse buccale (énanthème qui se caractérise par une langue framboisée, avec chéilite douloureuse (lèvres rouges et fissurées) ;
- une inflammation de l'épiderme ; l'exanthème (ou éruption cutanée) peut être de type scarlatiniforme, à type de grandes plaques confluentes, ou de type morbilliforme, par petites plaques, pouvant aussi ressembler à un syndrome de Stevens-Johnson. Elle s'accompagne d'une atteinte palmo-plantaire (érythème desquamatif pathognomonique, c'est-à-dire, signant la maladie) au 4e jour, avec œdème de la face dorsale des mains et des pieds. La desquamation apparaît vers la 2e ou 3e semaine d'évolution, au niveau de la jonction entre l'ongle et la pulpe des doigts et des orteils. L'éruption est particulièrement importante sur le site d'une précédente vaccination par le BCG, ce qui est très caractéristique de la maladie[3] ;
- un œdème (parfois) au niveau des mains et des pieds ;
- des ganglions gonflés au niveau du cou (adénopathies cervicales), souvent unilatéralement.
Examens complémentaires
Il existe un syndrome inflammatoire non spécifique avec hyperleucocytose, une VS augmentée, une augmentation de la CRP.
Parfois sont présentes une anémie, une thrombocytose, souvent tardive[4], ou une augmentation des immunoglobulines E.
Le diagnostic repose essentiellement sur les arguments cliniques (âge, tableau clinique et évolution), sur le syndrome inflammatoire biologique et sur la mise en évidence d'éventuels anévrismes coronariens à l'échocardiographie ou à la coronarographie [5].
Épidémiologie
Les pays asiatiques sont les plus touchés et particulièrement le Japon (avec environ 175 cas pour 100,000 enfants de moins de 5 ans chaque année). De plus, les Japonais vivants aux États-Unis ont une incidence de la maladie toujours aussi élevée que les Japonais vivant au Japon, suggérant fortement une origine génétique aux variations géographiques[6].
La maladie atteint, dans plus des trois quarts des cas, l'enfant entre 6 mois et 5 ans[3]. Elle est beaucoup plus fréquente au Japon où son incidence est un peu moins de 200 cas pour 100,000 enfants de moins de 5 ans[7]. Cette incidence est inférieure à 5 pour 100,000 enfants en Europe[8].
Cause
Ses causes sont restées totalement inconnues jusque dans les années 1990. On connaissait les particularités histologiques de la maladie, à savoir que les tissus atteints étaient infiltrés par des cellules immunitaires plutôt de type mononucléées, lymphocytes, macrophages, plasmocytes, mais assez peu de neutrophiles, alors que les neutrophiles sont les leucocytes les plus augmentés dans le sang lors de la maladie[9].
L'hypothèse infectieuse a été évoquée devant une prédominance hivernale[3] et la géographie groupée des cas. Une infection multi-bactérienne pourrait expliquer ce syndrome[10],[11] : la flore intestinale des malades contiendrait de 10 à 100 fois plus de deux bactéries pathogènes, des staphylocoques produisant des superantigènes auxquels le système immunitaire des malades répond par de la fièvre et d'éventuels œdèmes, et des bactéries caractérisées par une protéine dite « HSP 60 », qui stimulent également la production de cette protéine par les parois des vaisseaux sanguins qui sont alors attaqués par le système immunitaire du patient. La proportion de lymphocyte CD8+ dans les parois des artères atteintes est plus grande que la proportion de lymphocyte CD4+, ce qui suggérerait un germe intracellulaire[12].
La prédominance asiatique de la maladie ne diminuant pas lors de l'émigration[6] a fait émerger l'étude de potentiels facteurs génétiques qui rendrait plus susceptible de déclencher la maladie[13].
Toutefois, des simulations épidémiologiques orientent vers l'hypothèse d'une biotoxine préformée ou d'un agent environnemental aéroporté par des vents soufflant de la Chine du nord vers le Japon, non vers l'hypothèse infectieuse directe[14].
La piste aéroportée de l'origine de la maladie semble se confirmer : des spores infectieux du champignon Candida portés par les courant atmosphériques en seraient bien responsables [15]. En effet, En 2011, le climatologiste catalan Xavier Rodó découvre une corrélation entre les courants atmosphérique, le régime des vents, et les pics d'apparition de la maladie au Japon, à Hawaï et aux États-Unis[16].
Évolution et complications
La maladie évolue le plus souvent favorablement en deux à trois semaines (seuls 0,5 % à 2,8 % des malades en meurent), mais certains patients développent des complications cardiaques (anévrismes coronariens).
Le risque est cardiaque avec survenue d'une myocardite, d'une péricardite ou d'une atteinte des artères coronaires pouvant entraîner un infarctus, une insuffisance cardiaque aiguë et des troubles du rythme.
Une dilatation des artères coronaires peut être objectivée dans un peu moins de la moitié des cas dès la seconde semaine de la maladie[3]. Dans les quatre cinquièmes des cas, cette dilatation régresse mais des anévrismes peuvent apparaître dans le dernier cinquième. Dans ces cas, La cicatrisation se fait vers la constitution d'une fibrose de l'artère avec constitution de rétrécissements de cette dernière (sténose) se manifestant par un défaut d'apport d'oxygène au muscle cardiaque (ischémie), pouvant se compliquer d'angine de poitrine ou d'infarctus du myocarde. Ces complications surviennent, le plus souvent, moins d'un an après le début de la maladie[17].
D'autres artères peuvent avoir la même évolution de manière plus rare[3].
À long terme, le risque d'infarctus du myocarde persiste, même s'il est moindre[18].
Diagnostic différentiel
- Leptospirose ictéro-hémorragique ;
- Éruption allergique ;
- Maladie de Still (polyarthrite chronique juvénile) ;
- Érythème polymorphe mais absence de lésions muqueuses ;
- Scarlatine ;
- Rougeole ;
- Mononucléose infectieuse ;
- Collagénose.
Traitement
Il existe des recommandations publiées par l'American Heart Association en 2017, portant sur le diagnostic, la prise en charge et le traitement de la maladie de Kawasaki[19].
L'aspirine est utilisé comme anti-agrégant plaquettaire et comme anti-inflammatoire même si l'effet réel sur la maladie n'est pas formellement démontré[20].
Une cure unique d'immunoglobulines en intraveineuse à forte dose (2 g·kg-1 en deux jours) permet d'améliorer substantiellement l'état de l'enfant et réduit le risque de complications cardiaques[21].
En cas de récidives ou de formes résistantes, l'infliximab[22], les corticoïdes[23] ont été testés avec un certain succès.
Dans les formes graves, l'association d'immunoglobulines et de corticoïdes pourrait améliorer le pronostic cardiaque[24].
Surveillance
L'échographie du cœur permet de mesurer la dilatation des artères gauche et droite. Pour les enfants dont la maladie est survenue après l'âge de cinq ans, une épreuve d'effort peut être faite à partir de 6 ans.
Notes et références
- (en) Kawasaki T « Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children » Jpn J Allergy (Arerugi) 1967;16:178-222
- (en) Kim DS, « Kawasaki disease », Yonsei Medical Journal, vol. 47, no 6, , p. 759–72. (PMID 17191303, PMCID PMC2687814, DOI 10.3349/ymj.2006.47.6.759, lire en ligne)
- (en) Harnden A, Takahashi M, Burgner D, « Kawasaki disease » BMJ 2009;338:b1514. DOI:10.1136/bmj.b1514
- (en)Harnden A, Tulloh R, Burgner D, « Kawasaki disease » BMJ, 2014;349:g5336. DOI:10.1136/bmj.g5336
- Bressieux J-M, Rezzouk L et Soto B, « Kawasaki (maladie de) », sur www.therapeutique-dermatologique.org, (consulté le 20 juillet 2012)
- (en) Holman RC, Curns AT, Belay ED, Steiner CA, Schonberger LB, « Kawasaki syndrome hospitalizations in the United States, 1997 and 2000 », Pediatrics, vol. 112, no 3 Pt 1, , p. 495-501. (PMID 12949272)
- (en) Nakamura Y, Yashiro M, Uehara R, Oki I, Watanabe M, Yanagawa H, « Epidemiologic features of Kawasaki disease in Japan: results from the nationwide survey in 2005-2006 » J Epidemiol. 2008;18:167-72
- (en) Harnden A, Alves B, Sheikh A, « Rising incidence of Kawasaki disease in England: analysis of hospital admission data » BMJ 2002;324:1424-5
- (en) Amano S, Hazama F, Kubagawa H, Tasaka K, Haebara H, Hamashima Y, « General pathology of Kawasaki disease. On the morphological alterations corresponding to the clinical manifestations », Acta Pathol Jpn, vol. 30, no 5, , p. 681-94. (PMID 7446109)
- « Une infection par plusieurs bactéries à l'origine de la maladie de Kawasaki ? », http://www.bulletins-electroniques.com, le 4 décembre 2009.
- (en) Nagata S, Yamashiro Y, Ohtsuka Y, Shimizu T, Sakurai Y, Misawa S, Ito T, « Heat shock proteins and superantigenic properties of bacteria from the gastrointestinal tract of patients with Kawasaki disease », Immunology, vol. 128, no 4, , p. 511-20. (PMID 19950419, PMCID PMC2792135, DOI 10.1111/j.1365-2567.2009.03135.x, lire en ligne [html])
- (en) Brown TJ, Crawford SE, Cornwall ML, Garcia F, Shulman ST, Rowley AH, « CD8 T lymphocytes and macrophages infiltrate coronary artery aneurysms in acute Kawasaki disease », J Infect Dis, vol. 184, no 7, , p. 940-3. (PMID 11528596, DOI 10.1086/323155, lire en ligne [html])
- (en) Kyung Lim Yoon, « Update of genetic susceptibility in patients with Kawasaki disease », Korean J Pediatr, vol. 58, no 3, , p. 84-8. (PMID 25861330, PMCID PMC4388975, DOI 10.3345/kjp.2015.58.3.84, lire en ligne [PDF])
- (en) Rodó X, Curcoll R, Robinson M, Ballester J, Burns JC, Cayan DR, Lipkin WI, Williams BL, Couto-Rodriguez M, Nakamura Y, Uehara R, Tanimoto H, Morguí JA, « Tropospheric winds from northeastern China carry the etiologic agent of Kawasaki disease from its source to Japan », Proc Natl Acad Sci U S A, vol. 111, no 22, , p. 7952-7. (PMID 24843117, PMCID PMC4050536, DOI 10.1073/pnas.1400380111, lire en ligne)
- « A l'origine de la maladie de Kawasaki: un champignon transporté par le vent? » .
- (en) Mysterious disease rides on the winds across the Pacific Ocean » .
- (en) Kato H, Ichinose E, Kawasaki T, « Myocardial infarction in Kawasaki disease: clinical analyses in 195 cases » J Pediatr. 1986;108:923-7.
- (en) Kato H, Sugimura T, Akagi T, Sato N, Hashino K, Maeno Y et al. « Long-term consequences of Kawasaki disease. A 10- to 21-year follow-up study of 594 patients » Circulation 1996;94:1379-85.
- (en)McCrindle BW, Rowley AH, Newburger JW et al. Diagnosis, treatment, and long-term management of Kawasaki disease: A scientific statement for health professionals From the American Heart Association, Circulation, 2017;135:e927-e999
- (en) Baumer JH, Love SJ, Gupta A, Haines LC, Maconochie I, Dua JS, « Salicylate for the treatment of Kawasaki disease in children » Cochrane Database Syst Rev. 2006;(4):CD004175.
- (en) Oates-Whitehead RM, Baumer JH, Haines L, Love S, Maconochie IK, Gupta A et al. « Intravenous immunoglobulin for the treatment of Kawasaki disease in children » Cochrane Database Syst Rev. 2003;(4):CD004000.
- (en) Burns JC, Mason WH, Hauger SB et al. « Infliximab treatment for refractory Kawasaki syndrome » J Pediatr. 2005;146:662-667.
- (en) Wright DA, Newburger JW, Baker A, Sundel RP, « Treatment of immune globulin-resistant Kawasaki disease with pulsed doses of corticosteroids » J Pediatr. 1996;128:146-149.
- (en) Kobayashi T, Saji T, Otani T et al. on behalf of the RAISE study group investigators. « Efficacy of immunoglobulin plus prednisolone for prevention of coronary artery abnormalities in severe Kawasaki disease (RAISE study): a randomised, open-label, blinded-endpoints trial » Lancet 2012;379:1613-1620.
