Rhodocène
Le rhodocène ou bis(η5-cyclopentadiényl)rhodium(II), est un composé organométallique de la famille des métallocènes. De formule [Rh(C5H5)2], il est constitué d'un atome de rhodium lié par liaisons covalentes (haptiques) rhodium–carbone[3] à deux cycles de cyclopentadiényle entre lesquels il est pris en sandwich. Sa forme radicale est présente au-dessus de 150 °C ou piégée à des températures de type azote liquide (−196 °C). À température ambiante, des paires de ces radicaux se combinent pour former un dimère, ou deux des cycles de cyclopentadiényle sont joints, et se présentant sous la forme d'un solide jaune[1],[4],[5].
| Rhodocène | |

| |
| Identification | |
|---|---|
| Nom UICPA | bis(η5-cyclopentadienyl)rhodium(II) |
| Synonymes |
rhodocène |
| No CAS | |
| SMILES | |
| InChI | |
| Apparence | solide jaune (dimère)[1] |
| Propriétés chimiques | |
| Formule | C10H10Rh |
| Masse molaire[2] | 233,0919 ± 0,0087 g/mol C 51,53 %, H 4,32 %, Rh 44,15 %, |
| Propriétés physiques | |
| T° fusion | 174 °C (décomposition, dimère)[1] |
| Solubilité | légèrement dans le dichlorométhane (dimère)[1] soluble dans l'acétonitrile[1] |
| Composés apparentés | |
| Autres composés |
ferrocène, cobaltocène, iridocène, bis(benzène)chrome |
| Unités du SI et CNTP, sauf indication contraire. | |
L'histoire de la chimie organométallique inclut la découverte au XIXe siècle du sel de Zeise[6],[7] et la découverte par Ludwig Mond du tétracarbonyle de nickel[3]. Ces composés représentaient un défi aux chimistes, car ils ne pouvaient être décrits dans le modèle des liaisons chimiques d'alors. Un nouveau défi apparut à la découverte du ferrocène[8], l'analogue ferrique du rhodocène et premier composé connu de la famille des métallocènes[9]. Le ferrocène fut trouvé inhabituellement stable[10], comme l'étaient des structures chimiques analogues avec parmi elles le rhodocénium, le cation monopositif du rhodocène[Note 1], et ses équivalents au cobalt et à l'iridium[11]. L'étude de tels composés organométalliques eut pour conséquence le développement d'un nouveau modèle de liaisons, expliquant à la fois la formation de ces composés et leur stabilité[12],[13]. Les travaux sur les composés sandwich incluant le système rhodocénium / rhodocène valut aux chimistes Geoffrey Wilkinson et Ernst Otto Fischer le prix Nobel de chimie en 1973[14].
Du fait de leur stabilité et de la relative facilité à les préparer, les sels de rhodocénium sont des composés de départ usuels pour la préparation de rhodocène et de rhodocènes substitués, tous très instables. La synthèse originale utilise l'anion cyclopentadiénure et le tris(acétylacétonato)rhodium(III)[11] ; de nombreuses autres approches ont depuis été rapportées, incluant une transmétallation rédox en phase gaz[15] et utilisant des précurseurs demi-sandwich[16]. L'octaphénylrhodocène (un dérivé avec huit groupes phényle attachés) fut le premier rhodocène substitué à être isolé à température ambiante, même s'il se décompose rapidement dans l'air. La cristallographie aux rayons X a confirmé que l'octaphénylrhodocène a une structure de type sandwich dans une conformation décalée[17]. Contrairement au cobaltocène, qui est devenu un agent réducteur monoélectronique utile en recherche de laboratoire[18], aucun dérivé connu du rhodocène n'a une stabilité suffisante pour une quelconque application.
La recherche biomédicale a étudié les applications des composés du rhodium et de leurs dérivés en médecine[19] et a rapporté une application potentielle d'un dérivé du rhodocène comme produit radiopharmaceutique dans le traitement de petites tumeurs cancéreuses[20],[21]. Des dérivés du rhodocène sont aussi utilisés pour synthétiser des métallocènes liés, afin d'étudier les interactions métal–métal[22] ; des applications potentielles pour ces dérivés incluent l'électronique moléculaire et la recherche dans les mécanismes de catalyse[23]. La valeur des rhodocènes vient plutôt des informations qu'ils peuvent donner au sujet des modes de liaison et des dynamiques de nouveaux systèmes chimiques, que de leur utilisation directe dans des applications.
Histoire

Les découvertes en chimie organométallique ont permis d'obtenir des informations importantes sur la nature de la liaison chimique. Le sel de Zeise, K[PtCl3(C2H4)]·H2O, fut découvert en 1831[7] et Ludwig Mond découvrit Ni(CO)4 en 1888[25]. Chacun de ces composés contient une liaison entre un atome de métal et une petite molécule, l'éthylène dans les cas du sel de Zeise, et le dioxyde de carbone dans le cas du tétracarbonyle de nickel[6]. Le modèle compact de l'anion du sel de Zeise (à gauchce)[24] montre un lien direct entre l'atome de platine (en bleu) et l'atome de carbone (en noir) du ligand éthylène ; une telle liaison carbone-métal est la caractéristique d'un composé organométallique. Cependant, les modèles de liaison d'alors étaient incapables d'expliquer la nature de telles liaisons métal–alcène, jusqu'à ce que le modèle de Dewar-Chatt-Duncanson soit proposé dans les années 1950[12]. Sa formulation originale couverait seulement les liaisons métal–alcène[25], mais le modèle a été étendu par la suite pour décrire les systèmes de type carbonyle de métal (incluant [Ni(CO)4]) ou le système π est important[26].
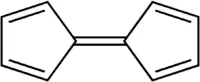


Le ferrocène, [Fe(C5H5)2], fut synthétisé la première fois en 1951 durant une tentative de préparer du fulvalène (bicyclopentadiénylidène, C10H8) par couplage oxydatif du cyclopentadiène ; le produit obtenu s'avéra avoir pour formule brute C10H10Fe, et il fut rapporté qu'il était d'une « remarquable stabilité »[10]. Cette découverte déclencha un vif intérêt dans le champ de la chimie organométallique[8],[9], en partie parce que la structure proposée par Pauson et Kealy (à droite) n'était pas compatible avec les modèles de liaison existant, et qu'elle n'expliquait pas cette stabilité inattendue. Ainsi, le premier défi fut de déterminer de façon certaine la structure du ferrocène dans l'espoir de comprendre son mode de liaison et ses propriétés. Trois groupes indépendants proposèrent la structure « sandwich » en 1952 : Robert Burns Woodward et Geoffrey Wilkinson étudièrent sa réactivité afin de déterminer sa structure[27] et démontrèrent que le ferrocène participe à des réactions similaires que des molécules aromatiques typiques (telles que le benzène)[28] ; Ernst Otto Fischer lui ne se contenta pas de déduire la structure sandwich, mais il commença à synthétiser d'autres métallocènes incluant le cobaltocène[29] ; Eiland et Pepinsky fournirent eux une confirmation par cristallographie aux rayons X de la structure sandwich[30]. L'application de la théorie de la liaison de valence au ferrocène, en considérant le centre Fe2+ et deux anions cyclopentadiénure (C5H5−), connus pour être aromatiques selon la règle de Hückel et donc hautement stable, permit une prédiction correcte de la géométrie de la molécule ; cependant, ce fut seulement lorsque la théorie de l'orbitale moléculaire fut appliquée avec succès que les raisons de la remarquable stabilité du ferrocène devinrent claires[13].
Les propriétés du cobaltocène rapportées par Wilkinson et Fischer démontrèrent que le cation monopositif cobalticinium [Co(C5H5)2]+ présentait une stabilité similaire à celle du ferrocène. Cette observation n'était pas inattendue du fait que le cation cobalticinium et le ferrocène sont isoélectroniques, même si le système de liaison n'était pas compris à cette époque. Quoi qu'il en soit, cette observation encouragea Wilkinson et Frank Albert Cotton à tenter de synthétiser les sels de rhodocénium[Note 1] et d'iridocénium[11]. Ils rapportèrent la synthèse de nombreux sels de rhodocénium, incluant le tribromure ([Rh(C5H5)2]Br3), le perchlorate ([Rh(C5H5)2]ClO4) et le reineckate ([Rh(C5H5)2] [Cr(NCS)4(NH3)2]·H2O), et trouvèrent que l'addition de dipicrylamine produisait un composé de formule [Rh(C5H5)2] [N(C6H2N3O6)2][11]. Dans chaque cas, le cation rhodocénium montra une grande stabilité. Wilkinson et Fischer partagèrent en 1973 le prix Nobel de chimie « pour leurs travaux de pionniers, réalisés indépendamment, sur les composés organométalliques appelés composés sandwich »[14].
La stabilité des métallocènes peut être directement comparée en regardant le potentiel d'oxydoréduction de la réduction monoélectronique de leur cation monopositif. Le tableau ci-dessous le présente avec pour référence l'électrode au calomel saturée (ECS) dans l'acétonitrile :
- [Fe(C5H5)2]+ / [Fe(C5H5)2] +0.38 V[31]
- [Co(C5H5)2]+ / [Co(C5H5)2] −0.94 V[1]
- [Rh(C5H5)2]+ / [Rh(C5H5)2] −1.41 V[1]
Ces données indiquent clairement la stabilité du ferrocène neutre et des cations cobaltocénium et rhodocénium. Le rhodocène est environ 500 mV plus réducteur que le cobaltocène, indiquant qu'il s'oxyde plus facilement et est donc moins stable[1]. Une étude plus ancienne par polarographie du perchlorate de rhodocénium à pH neutre montrait a cathodic wave peak⇔une onde cathodique avec un pic à −1,53 V (standard ECS) à l'électrode à gouttes tombantes de mercure, correspondant à la formation du rhodocène en solution ; cependant, les chercheurs furent incapables d'isoler le produit neutre de la solution. Dans la même étude, les tentatives de détecter l'iridocène en exposant les sels d'iridocénium à un milieu oxydant furent infructueuses à pH élevé[11]. Ces données sont cohérentes avec le fait que le rhodocène soit instable et pourraient indiquer que l'iridocène soit encore plus instable.
Description
La règle des 18 électrons est l'équivalent de la règle de l'octet pour la chimie des éléments du groupe principal, et est un outil pratique pour prédire la stabilité des composés organométalliques[32]. Elle permet de prédire que les espèces organométalliques dans lesquelles « la somme du nombre d'électrons de valence des éléments métalliques et de celui des électrons donnés par les ligands est de 18 ont de fortes chances d'être stables »[32]. Cette règle permet d'expliquer la forte stabilité inhabituelle du ferrocène[10] et des cations cobalticinium, rhodocénium[29] – ces trois espèces ayant des géométries analogues et étant isoélectroniques avec 18 électrons de valence. L'instabilité du rhodocène et du cobaltocène est aussi compréhensible via cette règle des 18 électrons, du fait que les deux composés ont 19 électrons de valence ; ceci peut expliquer les premières difficultés à isoler le rhodocène à partir des solutions de rhodocénium[11]. En fait, toute la chimie du rhodocène est dominée par ce besoin d'avoir une configuration à 18 électrons.
Le rhodocène existe sous la forme [Rh(C5H5)2], un monomère radical paramagnétique à 19 électrons seulement à une température inférieure ou égale à −196 °C (proche de la température d'ébullition de l'azote liquide), ou au-dessus de 150 °C, en phase gazeuse[1],[4],[5]. C'est cette forme qui présente la structure sandwich à conformation décalée typique. Cependant, à température ambiante (25 °C) la durée de vie de cette forme monomère dans l'acétonitrile est inférieure à deux secondes[1] ; en fait, le rhodocène forme un dimère [Rh(C5H5)2]2, avec une structure ansa-métallocène pontée diamagnétique avec 18 électrons de valence[33]. Des mesures par résonance paramagnétique électronique (RPE), résonance magnétique nucléaire (RMN) et spectroscopie infrarouge (IR) montrent l'existence d'un équilibre d'interconversion entre les formes monomère et dimère[5]. La RPE confirme également que le monomère possède un axe de symétrie de haut ordre (Cn, n > 2) avec un miroir plan (σ) perpendiculaire à ses éléments de symétrie ; ceci démontre expérimentalement que le monomère possède la structure typique sandwich des métallocènes[4],[Note 2] même si l'interprétation de ces données RPE est soumise à questions[33]. La voie de décomposition du monomère a aussi été étudiée par spectrométrie de masse[34]. La dimérisation est un processus redox, le dimère étant un composé du rhodium(I) alors que le monomère est un composé du rhodium(II)[Note 3]. Le rhodium occupe typiquement les états d'oxydation +I ou +III dans ses composés stables[35].

Le processus de dimérisation a pour effet global la diminution du nombre d'électrons autour de l'atome de rhodium central, de 19 à 18. Ceci se produit du fait du couplage oxydatif des deux ligands cyclopentadiényle produisant un nouveau ligand à plus faible hapticité et qui donne moins d'électrons au centre métallique. Ce terme d'hapticité est utilisé pour indiquer le « nombre d'atomes de carbone (ou autre) à travers lesquels [un ligand] se lie (n) »[36] à un centre métallique et est symbolisé par la notation ηn. Par exemple, le ligand éthylène du sel de Zeise est lié à l'atome de platine via ses deux atomes de carbone ; sa formule chimique est donc K[PtCl3(η2-C2H4)]·H2O. Les ligands carbonyle dans le tétracarbonyle de nickel sont tous liés via un seul atome de carbone et sont donc décrits comme des ligands monohaptiques, mais la notation η1 est en pratique omise de sa formule. Les ligands cyclopentadiényle dans la plupart des métallocènes et des composés demi-sandwich sont des ligands pentahaptiques, la formule chimique du rhodocène monomère est donc[Rh(η5-C5H5)2]. Dans le rhodocène dimérique, les ligands cyclopentadiényle couplés sont des donneurs de 4 électrons tétrahaptiques à chaque centre rhodium(I), et non pas donneurs de 6 électrons[Note 4] comme c'est le cas pour les donneurs cyclopentadiényle pentahaptiques. La stabilité accrue du dimère de rhodium(I) à 18 électrons de valence comparée au monomère de rhodium(II) à 19 électrons de valence explique en grande partie pourquoi le monomère n'est détecté que dans des conditions extrêmes[1],[5].

Cotton et Wilkinson ont montré[11] que le cation rhodocénium de rhodium(III) à 18 électrons de valence [Rh(η5-C5H5)2]+ peut être réduit en solution aqueuse à sa forme monomère ; cependant, ils furent incapables d'isoler le produit neutre, car non seulement il peut se dimériser, mais un monomère radical de rhodium(II) peut aussi spontanément former une espèce stable de rhodium(I) à hapticité mixte, [(η5-C5H5)Rh(η4-C5H6)][4]. Les différences entre le rhodocène et ses dérivées sont sur deux plans :
- l'une des liaisons des ligands cyclopentadiényle ont formellement gagné un atome d'hydrogène pour devenir un cyclopentadiène, qui reste lié au centre métallique mais pas comme un donneur de 4 électrons η4 ;
- le centre rhodium(II) est réduit en rhodium(I).
Ces deux changements font du dérivé une espèce à 18 électrons de valence. Fischer et ses collègues ont émis l'hypothèse que la formation de ce drivé de rhodocène pourrait se dérouler en deux étapes, une protonation, et une réduction, mais n'ont publié aucune preuve soutenant cette hypothèse[4]. (η4-Cyclopentadiène)(η5-cyclopentadienyl)rhodium(I), le composé résultant, est un complexe organométallique inhabituel du fait qu'il possède comme ligands à la fois l'anion cyclopentadiénure et le cyclopentadiène. Il a été montré que ce composé peut aussi être préparé par réduction au borohydrure de sodium d'une solution de rhodocénium solution dans l'éthanol aqueux ; les chercheurs qui ont fait cette découverte ont caractérisé le produit comme étant l'hydrure de biscyclopentadiénylrhodium[37].
Fischer et ses collègues ont aussi étudié la chimie de l'iridocène, l'analogue du rhodocène et du cobaltocène avec de l'iridium, troisième métal de transition dans la même colonne, trouvant que la chimie du rhodocène et de l'iridocène sont en général similaires. La synthèse de nombreux sels d'iridocénium incluant le tribromure et l'hexafluorophosphate ont été décrits[5]. Tout comme le rhodocène, l'iridocène se dimérise à température ambiante, mais la forme monomère peut être détectée à basse température et en phase gazeuse, et les mesures par IR, RMN et REP indiquent qu'il existe un équilibre chimique entre les deux formes, et confirment la structure sandwich du monomère iridocène[4],[5]. Le complexe [(η5-C5H5)Ir(η4-C5H6)], l'analogue du dérivé de rhodocène rapporté par Fischer[4], a aussi été étudié et montre des propriétés cohérentes avec le plus grand degré du système π du composé d'iridium(I) que celui de ses analogues au (I) et au rhodium(I)[38].
Synthèse
Les premiers sels de rhodocénium furent rapportés[11] deux ans après la découverte du ferrocène[10]. Ces sels furent préparés par réaction entre un carbanion de réactif de Grignard, le bromure de cyclopentadiénylmagnésium (C5H5MgBr) avec le tris(acétylacétonato)rhodium(III) (Rh(acac)3). Plus récemment, des cations rhodocénium en phase gazeuse ont été produits par transmétallation rédox d'ions rhodium(I) avec le ferrocène ou nickelocène[15].
- Rh+ + [(η5-C5H5)2M] → M + [(η5-C5H5)2Rh]+ M = Ni ou Fe
Des méthodes modernes de synthèse assistée par micro-ondes ont aussi été rapportées[39]. L'hexafluorophosphate de rhodocénium se forme par réaction entre le cyclopentadiène et le hydrate de chlorure de rhodium(III) dans le méthanol suivie d'une réaction avec l'hexafluorophosphate d'ammonium méthanolique ; cette réaction a un rendement dépassant 60 % avec seulement 30 secondes d'exposition aux micro-ondes[40].
- RhCl3.xH2O + 2 C5H6 + NH4PF6 → [(η5-C5H5)2Rh]PF6 + 2 HCl + NH4Cl + xH2O
Le rhodocène est lui formé par réduction de sels de rhodocénium avec du sodium fondu[4]. Si un produit de fusion contenant du rhodocénium est traité par le sodium ou le potassium métallique puis sublimé sur un support réfrigéré à l'azote liquide, un matériau noir polycristallin est formé[33]. En réchauffant ce matériau à température ambiante, on obtient un solide jaune dont on sait qu'il est un dimère de rhodocène. Une méthode similaire peut être utilisée pour préparer un dimère d'iridocène[33].
Rhodocènes substitués et sels de rhodocénium
Cation [(η5-C5tBu3H2)Rh(η5-C5H5)]+
De nouvelles approches en vue de synthétiser des complexes cyclopentadiényle substitués ont été développées en utilisant des vinylcyclopropènes substitués comme réactifs[41],[42],[43]. La réaction d'expansion de cycle de réarrangement du vinylcyclopropane (en) pour produire des cyclopentènes est bien connue[44] et ici utilisée pour réarranger les vinylcyclopropènes en cyclopentadiènes. Le cation [(η5-C5tBu3H2)Rh(η5-C5H5)]+ est produit par réaction en chaîne commençant par l'addition de dimère chlorobiséthylènerhodium(I) [(η2-C2H4)2Rh(μ-Cl)]2, sur le 1,2,3-tri-tert-butyl-3-vinyl-1-cyclopropène, suivie par une réaction avec le cyclopentadiénylthallium (en)[41],[42] :
Rh(C5H5))BF4.svg.png.webp)

Le pentadiénediyle de rhodium(III) à 18 électrons de valence produit par cette réaction montre encore l'instabilité de la partie rhodocène, au fait qu'il peut être chauffé à reflux dans le toluène pendant des mois sans que le 1,2,3-tri-tert-butylrhodocène ne se forme[pas clair], mais en conditions oxydantes, le cation 1,2,3-tri-tert-butylrhodocénium se forme rapidement[41]. La voltampérométrie cyclique a été utilisée pour étudier ce phénomène et d'autres similaires en détail[41],[42]. Il a été montré que le mécanisme de cette réaction implique la perte d'un électron par le ligand pentadiénediyle suivie par un réarrangement rapide (avec perte d'un atome d'hydrogène) pour former le cation 1,2,3-tri-tert-butylrhodocénium cation[42]. Les sels de tétrafluoroborate et d'hexafluorophosphate de ce cation ont été caractérisés par cristallographie aux rayons X[42].
[(η5-C5tBu3H2)Rh(η5-C5H5)]BF4 forme un cristal incolore centrosymétrique monoclinique appartenant au groupe d'espace P21/c avec une masse volumique de 1,486 g·cm−3[42]. Le diagramme ORTEP de la structure de ce cation (à droite) montre que ce composé a bien la géométrie attendue du rhodocène ou du cation rhodocénium. Les deux cycles de cyclopentadiényle sont presque parallèles (l'angle centroïde–Rh–centroïde est de 177,2°) et le centre de rhodium centre est légèrement plus près du cycle de cyclopentadiényle substitué (les distances Rh–centroïde sont de 1,819 Å et 1,795 Å), un fait attribué au plus grand effet inductif des groupes tert-butyle sur le ligand substitué[42]. Le diagramme ORTEP montre aussi que le cation adopte une conformation éclipsée à l'état solide. Cependant, la structure cristalline du sel d'hexafluorophosphate montre trois cations cristallographiquement indépendants : un est en conformation éclipsée ; l'autre en conformation décalée et le dernier est en rotation désordonnée[42]. Ceci laisse penser que la conformation adoptée est dépendante de l'anion présent et illustre aussi que la barrière énergétique à la rotation est basse – dans le ferrocène, cette barrière est d'environ 5 kJ·mol−1 aussi bien en solution qu'en phase gazeuse[13].
Rh(C5H5))BF4.png.webp)
Le diagramme ci-dessus montre les longueurs de liaison rhodium–carbone (en rouge, à l'intérieur des pentagones de gauche) et carbone–carbone (en bleu, à l'extérieur des pentagones, à gauche) pour les deux ligands, ainsi que les angles de liaison (en vert, dans les pentagones de droite) à l'intérieur de chaque cycle de cyclopentadiényle. La numérotation des atomes utilisée est la même que pour la structure cristalline. Pour le ligand cyclopentadiényle non-substitué, les longueurs de liaison carbone–carbone varient de 1,35 Å à 1,40 Å, et les angles internes varient de 107° à 109°. Par comparaison, dans un pentagone régulier, ces angles sont de 108°. Les longueurs de liaison rhodium–carbone varient elles de 2,16 Å à 2,18 Å[42]. Ces résultats sont cohérents avec la coordination η5 du ligand vers le centre métallique. Dans les cas du ligand cyclopentadiényle substitué, les variations sont plus grandes : les longueurs de liaison carbone–carbone varient de 1,39 Å à 1,48 Å, les angles internes de 106° à 111°, et les longueurs de liaison rhodium–carbone varient elles de 2,14 Å à 2,20 Å. Ces plus grandes variations dans les ligands substitués est attribuée aux distorsions nécessaires pour soulager le tension stérique imposée par les substituants tert-butyle voisins ; en dépit de ces variations, ces données montrent que le cyclopentadiényle substitué est aussi un ligand η5-coordonné[42].
La stabilité des métallocènes change en fonction de la substitution des cycles. En comparant les potentiels d'oxydoréduction des cations cobaltocénium et décaméthylcobaltocénium, on s'aperçoit que l'espèce décaméthylée est environ 600 mV moins réductrice que son parent métallocene[18], une situation aussi observée dans le cas du ferrocène[45] et du rhodocène[46]. Les données suivantes sont présentées de façon relative au couple rédox ferrocénium/ferrocène[47] :
| Demi-réaction | E° (V) |
|---|---|
| [Fe(C5H5)2]+ + e- ⇌ [Fe(C5H5)2] | 0 (par définition) |
| [Fe(C5Me5)2]+ + e- ⇌ [Fe(C5Me5)2] | −0.59[45] |
| [Co(C5H5)2]+ + e- ⇌ [Co(C5H5)2] | −1.33[18] |
| [Co(C5Me5)2]+ + e- ⇌ [Co(C5Me5)2] | −1.94[18] |
| [Rh(C5H5)2]+ + e- ⇌ [Rh(C5H5)2] | -1.79[1] † |
| [Rh(C5Me5)2]+ + e- ⇌ [Rh(C5Me5)2] | −2.38[46] |
| [(C5tBu3H2)Rh(C5H5)]+ + e- ⇌ [(C5tBu3H2)Rh(C5H5)] | -1.83[42] |
| [(C5tBu3H2)Rh(C5Me5)]+ + e- ⇌ [(C5tBu3H2)Rh(C5Me5)] | -2.03[42] |
| [(C5H5Ir(C5Me5)]+ + e- ⇌ [(C5H5Ir(C5Me5)] | -2.41[48] † |
| [Ir(C5Me5)2]+ + e- ⇌ [Ir(C5Me5)2] | -2.65[48] † |
| † après correction de 0,38 V[31] pour le standard différent |
Les différences de potentiel rédox dans le système cobaltocénium sont attribuées à l'effet inductif des groupes alkyle[18], stabilisant un peu plus l'espèce à 18 électrons de valence. Un effet similaire est observé avec les données du rhodocénium, encore une fois cohérente avec ces effets inductifs[42]. Dans le cas du système iridocénium substitué, les expériences de voltampérométrie cyclique ont montré des réductions irréversibles à des températures aussi basses que −60 °C[48] ; par comparaison, la réduction des rhodocènes correspondants est quasi-réversible à température ambiante et totalement réversible à −35 °C[46]. Cette irréversibilité des réductions d'iridocéniums substitués est attribuée à la dimérisation extrêmement rapide de l'espèce à 19 électrons de valence résultante, ce qui illustre une nouvelle fois que les iridocènes sont moins stables que les rhodocènes correspondants[48].
Ligands cyclopentadiényle penta-substitués
L'ensemble des connaissances sur les composés comportant des ligands cyclopentadiényle penta-substitués est vaste, et les plus connus de ces composés sont les complexes organométalliques avec les ligands pentaméthylcyclopentadienyle (Cp*) et pentaphénylcyclopentadiényle[49]. Les substitutions sur les cycles de cyclopentadiényle de rhodocènes et de sels de rhodocénium produisent des composés plus stables, car permettant une plus forte délocalisation de charges positives ou de densité électronique tout en provoquant un encombrement stérique empêchant d'autres espèces d'approcher le centre métallique[34]. De nombreuses espèces de rhodocénium mono- ou di-substituées sont connues, mais une stabilisation substantielle n'est obtenue qu'avec un plus grand nombre de substitutions[34]. Parmi ces sels de rhodocénium hautement substitués, on compte l'hexafluorophosphate de décaméthylrhodocénium [(η5-C5Me5)2Rh]PF6[50], l'hexafluorophosphate de décaisopropylrhodocénium [(η5-C5iPr5)2Rh]PF6[51], et l'hexafluorophosphate d'octaphénylrhodocénium [(η5-C5Ph4H)2Rh]PF6[17],[Note 5]. Le tétrafluoroborate de décaméthylrhodocénium peut être synthétisé à partir du complexe de tris(acétone) [(η5-C5Me5)Rh(Me2CO)3](BF4)2 par réaction avec le pentaméthylcyclopentadiène, la même méthode fonctionnant avec l'analogue d'iridium[52]. L'hexafluorophosphate de décaisopropylrhodocénium a été synthétisé dans le diméthoxyéthane (solvant) par une inhabituelle synthèse monotope impliquant la formation de 20 liaisons carbone-carbone[51] :

Dans une réaction similaire, l'hexafluorophosphate de pentaisopropylrhodocénium [(η5-C5iPr5)Rh(η5-C5H5)]PF6 peut être synthétysé à partir de l'hexafluorophosphate de pentaméthylrhodocénium [(η5-C5Me5)Rh(η5-C5H5)]PF6 avec un rendement de 80 %[51]. Ces réactions montrent que l'acidité des hydogènes des groupes méthyle du complexe de pentaméthylcyclopentadiényle peut être considérablement accrue par la présence d'un centre métallique. Le mécanisme de cette réaction consiste en une déprotonation d'un groupe méthyle par l'hydroxyde de potassium résultant en un carbanion qui subit ensuite une substitution nucléophile avec l'iodométhane pour former une nouvelle liaison carbone-carbone[51].
Les composés tétrafluoroborate de pentaphénylrhodocénium [(η5-C5Ph5)Rh(η5-C5H5)]BF4 et tétrafluoroborate de pentaméthylpentaphénylrhodocénium [(η5-C5Ph5)Rh(η5-C5Me5)]BF4 ont aussi été étudiés. Ils ont permis de montrer qu'il était possible de préparer des composés sandwich du rhodium à partir de précurseurs demi-sandwich. Par exemple, dans une approche globalement similaire à la synthèse du tétrafluoroborate décaméthylrhodocénium partir de tris(acétone)[52], le tétrafluoroborate de pentaphénylrhodocénium a été synthétisé à partir du sel de tris(acétonitrile) [(η5-C5Ph5)Rh(CH3CN)3](BF4)2 par réaction avec le cyclopentadiénure de sodium[16] :
- [(η5-C5Ph5)Rh(MeCN)3](BF4)2 + NaC5H5 → [(η5-C5Ph5)Rh(η5-C5H5)]BF4 + NaBF4 + 3 MeCN

L'octaphénylrhodocène, [(η5-C5Ph4H)2Rh], est le premier dérivé de rhodocène à avoir été isolé à température ambiante. Il se présente sous la forme de cristaux vert olive se décomposant rapidement en solution, et en quelques minutes à l'air libre, montrant une sensibilité à l'air considérablement plus grande que son analogue au cobalt, même s'il est significativement plus stable que le rhodocène lui-même. Cette différence est attribuée à la stabilité relative du rhodium(II), plus faible que celle du cobalt(II)[17],[35]. Le potentiel rédox du cation [(η5-C5Ph4H)2Rh]+ (mesurée dans le diméthylformamide par rapport au couple ferrocénium/ferrocène) et de −1,44 V, cohérent avec la plus grande stabilisation thermodynamique du rhodocène par le ligand C5HPh4, comparé aux ligands C5H5 our C5Me5[17]. Le cobaltocène est un agent réducteur monoélectronique utile en recherche en laboratoire car il est soluble dans les solvants apolaires organiques[18] et son couple rédox se comporte suffisamment bien pour être utilisé comme standard interne en voltampérométrie cyclique[53]. Aucun rhodocène substitué préparé à ce jour n'a montré de stabilité suffisante pour être utilisé de façon similaire.
La synthèse de l'octaphénylrhodocènese se fait en trois étapes, un reflux avec le diglyme suivi d'une réaction avec l'acide hexafluorophosphorique, et enfin une réduction par un amalgame de sodium dans le tétrahydrofurane[17] :
- Rh(acac)3 + 2 KC5Ph4H → [(η5-C5Ph4H)2Rh]+ + 2 K+ + 3 acac−
- [(η5-C5Ph4H)2Rh]+ + 3 acac− + 3 HPF6 → [(η5-C5Ph4H)2Rh]PF6 + 3 Hacac + 2 PF6−
- [(η5-C5Ph4H)2Rh]PF6 + Na/Hg → [(η5-C5Ph4H)2Rh] + NaPF6
La structure cristalline obtenue par cristallographie aux rayons X montre que l'octaphénylrhodocène adopte une conformation décalée[17], similaire à celle du ferrocène, et opposée à la conformation éclipsée du ruthénocène[13]. La distance rhodium–centroïde et de 1,904 Å, les longueurs de liaison rhodium–carbone sont en moyenne de 2,26 Å et les liaisons carbone–carbone en moyenne de 1,44 Å[17]. Ces distances sont toutes similaires à celles du cation 1,2,3-tri-tert-butylrhodocénium décrit plus haut, la seule différence étant la taille effective du centre rhodium qui apparaît plus grand, une observation cohérente avec le fait que le rayon ionique du rhodium(II) soit plus grand que celui du rhodium(III).
Applications
Utilisation biomédicale de dérivés

Les composés métallopharmaceutiques font l'objet d'une vaste étude[54],[55], parmi eux certains composés du rhodium[19], et en particulier ses dérivés métallocènes[56], tous comme les dérivés du ferrocène[57]. Un point d'intérêt particulier est l'utilisation de métallocènes à la place du groupe fluorophényle dans l'haloperidol[20], un antipsychotique typique. Le composé ferrocényl–halopéridol étudié a pour structure (C5H5)Fe(C5H4)–C(=O)–(CH2)3–N(CH2CH2)2C(OH)–C6H4Cl et peut être converti en son analogue au ruthénium par transmétallation. Em utilisant le radioisotope 103Ru, on a ainsi pu produire un ruthénocényl–halopéridol, un composé radiopharmaceutique avec une forte affinité pour les tissus du poumon mais pas ceux du cerveau chez la souris et le rat[20]. La désintégration β de 103Ru produit un isomère nucléaire métastable, le rhodium 103m (103mRh), et donne le composé rhodocényl–halopéridol. Ce composé, comme d'autres dérivés du rhodocène, a une configuration électronique instable avec 19 électrons de valence, et s'oxyde rapidement en rhodocénium–halopéridol cationique voulu[20]. La séparation du ruthénocényl–halopéridol et du rhodocénium–halopéridol et leur distribution respective dans les organes a été étudiée[21]. 103mRh a une demi-vie de 56 minutes et émet une radiation gamma de 39,8 keV ; ainsi, la désintégration gamma de l'isotope du rhodium suit assez rapidement la désintégration bêta de l'isotope du ruthénium. Il a été proposé d'utiliser les radioisitopes iode 131 (131I), fer 59 (59Fe), calcium 47 (47Ca) et 103mRh comme émetteurs de radiations bêta et gamma pour traiter les petites tumeurs par radiothérapie[19].
Interactions métal–métal dans les métallocènes liés

La motivation initiale des études du système rhodocène était de comprendre la nature des liaisons à l'intérieur des métallocènes. Plus récemment, l'intérêt a été ravivé par le désir d'étudier les interactions métal-métal dans les métallocènes liés[22]. Les applications potentielles de tels systèmes incluent l'électronique moléculaire[23] , les polymères métallocènes semi-conducteurs (et possiblement ferromagnétiques)[22] ces derniers pouvant être des exemples de fil moléculaire (en), et l'exploration de la frontière entre la catalyse hétérogène et la catalyse homogène[23]. Parmi les bimétallocènes et termétallocènes connus possédant une partie de rhodocényle, on compte les sels d'hexafluorophosphate du rhodocénylferrocène, 1,1'-dirhodocénylferrocène, et 1-cobaltocényl-1'-rhodocénylferrocène[58]. Des métallocènes liés peuvent aussi être formés en introduisant divers substituants métallocényle sur un seul ligand cyclopentadiényle[23].
L'étude structurelle des termétallocènes montre qu'ils adoptent typiquement une géométrie "eclipsed double transoid" "crankshaft"⇔« vilebrequin » « double transoïde eclipsé »[59]. En prenant l'exemple du cation 1-cobaltocényl-1'-rhodocénylferrocène illustré ci-dessus, cela signifie que les parties cobaltocényle et rhodocényle sont en conformation éclipsée, et donc que les atomes de carbone 1 et 1' sur le ferrocène central (les atomes de carbone de chacun des cyclopentadiényles du ferrocène liés respectivement au cyclopentadiényle bas de la partie rhodocène et au cyclopentadiényle haut de la partie cobaltocène) sont aussi verticalement alignés que possible, du fait de la confirmation décalée de cycles de cyclopentadiényle à l'intérieur de chaque unité de métallocène. Vu de côté, cela signifie que les termétallocènes ressemblent à la forme bas-haut-bas d'un vilebrequin[59]. La synthèse de ces termétallocènes implique le mélange de solutions de rhodocénium et de cobaltocénium avec le 1,1'-dilithioferrocène. Ceci produit un intermédiaire non-chargé avec des ligands cyclopentadiényle–cyclopentadiène liés dont les liaisons ressemblent à celles trouvées dans les dimères de rhodocène. Ces ligands réagissent ensuite avec un carbocation triphénylméthyle (en) pour produire le sel de termétallocène, [(η5-C5H5)Rh(μ-η5:η5-C5H4–C5H4)Fe(μ-η5:η5-C5H4–C5H4)Co(η5-C5H5)](PF6)2. Cette synthèse est illustrée ci-dessous[58],[59] :

Notes et références
Notes
- Le cation [Rh(C5H5)2]+ avec 18 électrons de valence est appelé cation rhodocénium dans certains articles[1], mais cation rhodicinium dans d'autres[11]. La première forme est la plus courante dans les articles récents, et c'est celle adopté dans cet article, mais ces deux noms décrivent bien à la même espèce chimique
- La présence d'une miroir plan perpendiculaire à l'axe de symétrie centroïde du cycle C5 –métal–centroïde suggère une conformation éclipsée plutôt qu'une conformation décalée. Cependant, la rotation libre des ligands cyclopentadiényle autour de cet axe est commune chez les métallocènes – dans le ferrocène, la barrière énergétique de cette rotation est d'environ 5 kJ·mol−1[13]. En conséquence, des formes monomères du rhodocène en conformation éclipsée et en conformation décalée co-existeraient, et s'interconvertirait rapidement en solution. C'est seulement à l'état solide qu'une conformation éclipsée ou décalée définitive à réellement du sens.
- Dans le rhodocène dimérique, les cycles joints de cyclopentadiène sont représentés avec les atomes d'hydrogène en position endo (c'est-à-dire que les H sont à l'intérieur l'autre moitié des ligands est à l'extérieur.). Même si ce n'est pas basé sur les données de structure cristalline, cela suit les illustrations fournies par El Murr et al.[1] et par Fischer et Wawersik[4] dans la discussion sur les données RMNH qu'ils obtenu. L'article de Collins et al.[17], montre cependant les atomes d;hydrogène en position exo.
- Il existe deux approches distinctes pour le comptage d'électron, basés soit sur l'espèce radicale, soit sur l'espèce ionique. En utilisant l'approche « radical », le centre de rhodium a 9 électrons, quel que soit son état d'oxydation,et le ligand cyclopentadiényle est donneur de 5 électrons. En utilisant l'approche « ion », le ligand cyclopentadiényle est un donneur de 6 électrons et le compte d'électrons du centre de rhodium dépend de son état d'oxydation – le rhodium(I) est un centre à 8 électrons, le rhodium(II) est un centre à 7 électrons, et le rhodium(III) est un centre à 6 électrons. Les deux approches donnent en général le même résultat mais il est important d'être cohérent en utilisant soit l'une, soit l'autre.
- Parmi les abréviations courantes de fragments de molécule, « Me » désigne le groupe méthyle, —CH3 ; « iPr » désigne le groupe iso-propyle, —CH(CH3)2 ; « Ph » désigne le groupe phényle, —C6H5 ; « tBu » désigne le groupe tert-butyle, —C(CH3)3.
Références
- N. El Murr, J. E. Sheats, W. E. Geiger et J. D. L. Holloway, « Electrochemical Reduction Pathways of the Rhodocenium Ion. Dimerization and Reduction of Rhodocene », Inorg. Chem., vol. 18, no 6, , p. 1443–1446 (DOI 10.1021/ic50196a007)
- Masse molaire calculée d’après « Atomic weights of the elements 2007 », sur www.chem.qmul.ac.uk.
- (en) R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, Hoboken, NJ, John Wiley and Sons, , 5e éd., 505 p. (ISBN 978-0-470-25762-3, lire en ligne), p. 2
« An industrial application of transition metal organometallic chemistry appeared as early as the 1880s, when Ludwig Mond showed that nickel can be purified by using CO to pick up nickel in the form of gaseous Ni(CO)4 that can easily be separated from solid impurities and later be thermally decomposed to give pure nickel.
... Recent work has shown the existence of a growing class of metalloenzymes having organometallic ligand environments – considered as the chemistry of metal ions having C-donor ligands such as CO or the methyl group »
- (de) E. O. Fischer et H. Wawersik, « Über Aromatenkomplexe von Metallen. LXXXVIII. Über Monomeres und Dimeres Dicyclopentadienylrhodium und Dicyclopentadienyliridium und Über Ein Neues Verfahren Zur Darstellung Ungeladener Metall-Aromaten-Komplexe », J. Organomet. Chem., vol. 5, no 6, , p. 559–567 (DOI 10.1016/S0022-328X(00)85160-8)
- (de) H. J. Keller et H. Wawersik, « Spektroskopische Untersuchungen an Komplexverbindungen. VI. EPR-spektren von (C5H5)2Rh und (C5H5)2Ir », J. Organomet. Chem., vol. 8, no 1, , p. 185–188 (DOI 10.1016/S0022-328X(00)84718-X)
- L. B. Hunt, « The First Organometallic Compounds: William Christopher Zeise and his Platinum Complexes », Platinum Metals Rev., vol. 28, no 2, , p. 76–83 (lire en ligne)
- (de) W. C. Zeise, « Von der Wirkung zwischen Platinchlorid und Alkohol, und von den dabei entstehenden neuen Substanzen », Ann. der Physik, vol. 97, no 4, , p. 497–541 (DOI 10.1002/andp.18310970402, Bibcode 1831AnP....97..497Z)
- P. Laszlo et R. Hoffmann, « Ferrocene: Ironclad History or Rashomon Tale? », Angew. Chem. Int. Ed., vol. 39, no 1, , p. 123–124 (PMID 10649350, DOI 10.1002/(SICI)1521-3773(20000103)39:1<123::AID-ANIE123>3.0.CO;2-Z)
- A. Federman Neto, A. C. Pelegrino et V. A. Darin, « Ferrocene: 50 Years of Transition Metal Organometallic Chemistry — From Organic and Inorganic to Supramolecular Chemistry », ChemInform, vol. 35, no 43, (DOI 10.1002/chin.200443242) (Abstract; original published in Trends Organomet. Chem., 4:147–169, 2002)
- T. J. Kealy et P. L. Pauson, « A New Type of Organo-Iron Compound », Nature, vol. 168, no 4285, , p. 1039–1040 (DOI 10.1038/1681039b0, Bibcode 1951Natur.168.1039K)
- F. A. Cotton, R. O. Whipple et G. Wilkinson, « Bis-Cyclopentadienyl Compounds of Rhodium(III) and Iridium(III) », J. Am. Chem. Soc., vol. 75, no 14, , p. 3586–3587 (DOI 10.1021/ja01110a504)
- D. M. P. Mingos, « A Historical Perspective on Dewar's Landmark Contribution to Organometallic Chemistry », J. Organomet. Chem., vol. 635, nos 1–2, , p. 1–8 (DOI 10.1016/S0022-328X(01)01155-X)
- (en) Mehrotra, R. C. et Singh, A., Organometallic Chemistry : A Unified Approach, New Delhi, New Age International, , 2e éd., 632 p. (ISBN 978-81-224-1258-1, lire en ligne), p. 261–267
- « The Nobel Prize in Chemistry 1973 », Nobel Foundation (consulté le )
- D. B. Jacobson, G. D. Byrd et B. S. Freiser, « Generation of Titanocene and Rhodocene Cations in the Gas Phase by a Novel Metal-Switching Reaction », J. Am. Chem. Soc., vol. 104, no 8, , p. 2320–2321 (DOI 10.1021/ja00372a041)
- (en) H. T. He, Synthesis and Characterisation of Metallocenes Containing Bulky Cyclopentadienyl Ligands (PhD thesis), University of Sydney, (OCLC 222646266)
- J. E. Collins, M. P. Castellani, A. L. Rheingold, E. J. Miller, W. E. Geiger, A. L. Rieger et P. H. Rieger, « Synthesis, Characterization, and Molecular-Structure of Bis(tetraphenylcyclopentadienyl)rhodium(II) », Organometallics, vol. 14, no 3, , p. 1232–1238 (DOI 10.1021/om00003a025)
- N. G. Connelly et Geiger, « Chemical Redox Agents for Organometallic Chemistry », Chem. Rev., vol. 96, no 2, , p. 877–910 (PMID 11848774, DOI 10.1021/cr940053x)
- (en) F. P. Pruchnik, Metallotherapeutic Drugs and Metal-Based Diagnostic Agents : The Use of Metals in Medicine, Hoboken, NJ, Wiley, , 598 p. (ISBN 0-470-86403-6, DOI 10.1002/0470864052.ch20, lire en ligne), « 45Rh — Rhodium in Medicine », p. 379–398
- (de) M. Wenzel et Y. Wu, « Ferrocen-, Ruthenocen-bzw. Rhodocen-analoga von Haloperidol Synthese und Organverteilung nach Markierung mit 103Ru-bzw. 103mRh », Int. J. Rad. Appl. Instrum. A., vol. 39, no 12, , p. 1237–1241 (PMID 2851003, DOI 10.1016/0883-2889(88)90106-2)
- (de) M. Wenzel et Y. F. Wu, « Abtrennung von [103mRh]Rhodocen-Derivaten von den Analogen [103Ru]Ruthenocen-Derivaten und deren Organ-Verteilung », Int. J. Rad. Appl. Instrum. A., vol. 38, no 1, , p. 67–69 (PMID 3030970, DOI 10.1016/0883-2889(87)90240-1)
- S. Barlow et D. O'Hare, « Metal–Metal Interactions in Linked Metallocenes », Chem. Rev., vol. 97, no 3, , p. 637–670 (DOI 10.1021/cr960083v)
- M. Wagner, « A New Dimension in Multinuclear Metallocene Complexes », Angew. Chem. Int. Ed., vol. 45, no 36, , p. 5916–5918 (DOI 10.1002/anie.200601787)
- J. A. J. Jarvis, B. T. Kilbourn et P. G. Owston, « A Re-determination of the Crystal and Molecular Structure of Zeise's salt, KPtCl3.C2H4.H2O », Acta Cryst. B, vol. 27, no 2, , p. 366–372 (DOI 10.1107/S0567740871002231)
- (en) Modern Coordination Chemistry : The Legacy of Joseph Chatt, Cambridge, UK, RSC Publishing, , 386 p. (ISBN 0-85404-469-8, lire en ligne), « Section D: Transition Metal Complexes of Olefins, Acetylenes, Arenes and Related Isolobal COmpounds », p. 101–110
- (en) D. Astruc, Organometallic Chemistry and Catalysis, Berlin, Springer, , 608 p. (ISBN 978-3-540-46128-9, lire en ligne), p. 41–43
- G. Wilkinson, M. Rosenblum, M. C. Whiting et R. B. Woodward, « The Structure of Iron Bis-Cyclopentadienyl », J. Am. Chem. Soc., vol. 74, no 8, , p. 2125–2126 (DOI 10.1021/ja01128a527)
- (en) H. Werner, Landmarks in Organo-Transition Metal Chemistry : A Personal View, New York, Springer Science, (ISBN 978-0-387-09847-0, lire en ligne), p. 161–163
- (de) E. O. Fischer et W. Pfab, « Zur Kristallstruktur der Di-Cyclopentadienyl-Verbindungen des zweiwertigen Eisens, Kobalts und Nickels », Z. Anorg. Allg. Chem., vol. 7, no 6, , p. 377–379 (DOI 10.1002/zaac.19532740603)
- P. F. Eiland et R. Pepinsky, « X-ray Examination of Iron Biscyclopentadienyl », J. Am. Chem. Soc., vol. 74, no 19, , p. 4971 (DOI 10.1021/ja01139a527)
- V. V. Pavlishchuk et A. W. Addison, « Conversion Constants for Redox Potentials Measured Versus Different Reference Electrodes in Acetonitrile Solutions at 25 °C », Inorg. Chim. Acta, vol. 298, no 1, , p. 97–102 (DOI 10.1016/S0020-1693(99)00407-7)
- (en) J. C. Kotz, P. M. Treichel et J. R. Townsend, Chemistry and Chemical Reactivity, Volume 2, Belmont, CA, Cengage Learning, , 7e éd., 1312 p. (ISBN 978-0-495-38703-9, lire en ligne), p. 1050–1053
- B. De Bruin, D. G. H. Hetterscheid, A. J. J. Koekkoek et H. Grützmacher, « The Organometallic Chemistry of Rh–, Ir–, Pd–, and Pt–Based Radicals: Higher Valent Species », Prog. Inorg. Chem., vol. 55, , p. 247–354 (ISBN 978-0-471-68242-4, DOI 10.1002/9780470144428.ch5, lire en ligne)
- D. V. Zagorevskii et J. L. Holmes, « Observation of Rhodocenium and Substituted-Rhodocenium Ions and their Neutral Counterparts by Mass Spectrometry », Organometallics, vol. 11, no 10, , p. 3224–3227 (DOI 10.1021/om00046a018)
- (en) S. A. Cotton, Chemistry of Precious Metals, Londres, Blackie Academic and Professional, (ISBN 0-7514-0413-6, lire en ligne), « Rhodium and Iridium », p. 78–172
« Both metals exhibit an extensive chemistry, principally in the +3 oxidation state, with +1 also being important, and a significant chemistry of +4 iridium existing. Few compounds are known in the +2 state, in contrast to the situation for cobalt, their lighter homologue (factors responsible include the increased stability of the +3 state consequent upon the greater stabilization of the low spin d6 as 10 Dq increases)." (p. 78) »
- (en) A. F. Hill, Organotransition Metal Chemistry, Cambridge, UK, Royal Society of Chemistry, , 185 p. (ISBN 0-85404-622-4, lire en ligne), p. 4–7
- M. L. H. Green, L. Pratt et G. Wilkinson, « 760. A New Type of Transition Metal–Cyclopentadiene Compound », J. Chem. Soc., , p. 3753–3767 (DOI 10.1039/JR9590003753)
- L. P. Szajek et J. R. Shapley, « Unexpected Synthesis of CpIr(η4-C5H6) and a Proton and Carbon-13 NMR Comparison with its Cobalt and Rhodium Congeners », Organometallics, vol. 10, no 7, , p. 2512–2515 (DOI 10.1021/om00053a066)
- D. R. Baghurst et D. M. P. Mingos, « Design and Application of a Reflux Modification for the Synthesis of Organometallic Compounds Using Microwave Dielectric Loss Heating Effects », J. Organomet. Chem., vol. 384, no 3, , C57–C60 (DOI 10.1016/0022-328X(90)87135-Z)
- D. R. Baghurst, D. M. P. Mingos et M. J. Watson, « Application of Microwave Dielectric Loss Heating Effects for the Rapid and Convenient Synthesis of Organometallic Compounds », J. Organomet. Chem., vol. 368, no 3, , C43–C45 (DOI 10.1016/0022-328X(89)85418-X)
- B. T. Donovan-Merkert, H. I. Tjiong, L. M. Rhinehart, R. A. Russell et J. Malik, « Facile, Redox-Promoted Formation of Rhodocenium Complexes Bearing the 1,2,3-Tri-tert-butylcyclopentadienyl Ligan », Organometallics, vol. 16, no 5, , p. 819–821 (DOI 10.1021/om9608871)
- B. T. Donovan-Merkert, C. R. Clontz, L. M. Rhinehart, H. I. Tjiong, C. M. Carlin, Thomas R. Cundari, Arnold L. Rheingold et Ilia Guzei, « Rhodocenium Complexes Bearing the 1,2,3-Tri-tert-butylcyclopentadienyl Ligand: Redox-Promoted Synthesis and Mechanistic, Structural and Computational Investigations », Organometallics, vol. 17, no 9, , p. 1716–1724 (DOI 10.1021/om9707735)
- Hughes, R. P., Trujillo, H. A., Egan, J. W. et Rheingold, A. L., « Skeletal Rearrangement during Rhodium-Promoted Ring Opening of 1,2-Diphenyl-3-vinyl-1-cyclopropene. Preparation and Characterization of 1,2- and 2,3-Diphenyl-3,4-pentadienediyl Rhodium Complexes and Their Ring Closure to a 1,2-Diphenylcyclopentadienyl Complex », Organometallics, vol. 18, no 15, , p. 2766–2772 (DOI 10.1021/om990159o)
- Z. Goldschmidt et B. Crammer, « Vinylcyclopropane Rearrangements », Chem. Soc. Rev., vol. 17, , p. 229–267 (DOI 10.1039/CS9881700229)
- I. Noviandri, K. N. Brown, D. S. Fleming, P. T. Gulyas, P. A. Lay, A. F. Masters et L. Phillips, « The Decamethylferrocenium/Decamethylferrocene Redox Couple: A Superior Redox Standard to the Ferrocenium/Ferrocene Redox Couple for Studying Solvent Effects on the Thermodynamics of Electron Transfer », J. Phys. Chem. B, vol. 103, no 32, , p. 6713–6722 (DOI 10.1021/jp991381)
- O. V. Gusev, L. I. Denisovich, M. G. Peterleitner, A. Z. Rubezhov, Nikolai A. Ustynyuk et P. M. Maitlis, « Electrochemical Generation of 19- and 20-electron Rhodocenium Complexes and Their Properties », J. Organomet. Chem., vol. 452, nos 1–2, , p. 219–222 (DOI 10.1016/0022-328X(93)83193-Y)
- Gagne, R. R., Koval, C. A. et Lisensky, G. C., « Ferrocene as an Internal Standard for Electrochemical Measurements », Inorg. Chem., vol. 19, no 9, , p. 2854–2855 (DOI 10.1021/ic50211a080)
- O. V. Gusev, M. G. Peterleitner, M. A. Ievlev, A. M. Kal'sin, P. V. Petrovskii, L. I. Denisovich et Nikolai A. Ustynyuk, « Reduction of Iridocenium Salts [Ir(η5-C5Me5)(η5-L)]+ (L= C5H5, C5Me5, C9H7); Ligand-to-Ligand Dimerisation Induced by Electron Transfer », J. Organomet. Chem., vol. 531, nos 1–2, , p. 95–100 (DOI 10.1016/S0022-328X(96)06675-2)
- (en) J. Okuda, Transition Metal Coordination Chemistry, vol. 160, Berlin, Springer-Verlag, coll. « Topics in Current Chemistry », , 148 p. (ISBN 3-540-54324-4, DOI 10.1007/3-540-54324-4_3), « Transition-Metal Complexes of Sterically Demanding Cyclopentadienyl Ligands », p. 97–145
- (de) U. Kölle et W. Z.l Kläui, « Darstellung und Redoxverhalten einer Serie von Cp*/aqua/tripod-Komplexen des Co, Rh und Ru », Z. Naturforsch. B, vol. 46, no 1, , p. 75–83
- D. Buchholz et D. Astruc, « The First Decaisopropylmetallocene – One-Pot Synthesis of [Rh(C5iPr5)2]PF6 from [Rh(C5Me5)2]PF6 by Formation of 20 Carbon–Carbon Bonds », Angew. Chem. Int. Ed., vol. 33, nos 15–16, , p. 1637–1639 (DOI 10.1002/anie.199416371)
- Gusev, O. V., Morozovaa, L. N., Peganovaa, T. A., Petrovskiia, P. V., Ustynyuka N. A. et Maitlis, P. M., « Synthesis of η5-1,2,3,4,5-Pentamethylcyclopentadienyl-Platinum Complexes », J. Organomet. Chem., vol. 472, nos 1–2, , p. 359–363 (DOI 10.1016/0022-328X(94)80223-8)
- R. S. Stojanovic et A. M. Bond, « Examination of Conditions under which the Reduction of the Cobaltocenium Cation can be used as a Standard Voltammetric Reference Process in Organic and Aqueous Solvents », Anal. Chem., vol. 65, no 1, , p. 56–64 (DOI 10.1021/ac00049a012)
- (en) Clarke, M. J. et Sadler, P. J., Metallopharmaceuticals : Diagnosis and therapy, Berlin, Springer, (ISBN 3-540-65308-2)
- (en) C. J. Jones et J. Thornback, Medicinal Applications of Coordination Chemistry, Cambridge, UK, RSC Publishing, , 353 p. (ISBN 978-0-85404-596-9, lire en ligne)
- M. J. Clarke, « Ruthenium Metallopharmaceuticals », Coord. Chem. Rev., vol. 232, nos 1–2, , p. 69–93 (DOI 10.1016/S0010-8545(02)00025-5)
- M. F. R. Fouda, M. M. Abd-Elzaher, R. A. Abdelsamaia et A. A. Labib, « On the Medicinal Chemistry of Ferrocene », Appl. Organomet. Chem., vol. 21, no 8, , p. 613–625 (DOI 10.1002/aoc.1202)
- M. Andre, H. Schottenberger, R. Tessadri, G. Ingram, P. Jaitner et K. E. Schwarzhans, « Synthesis and Preparative HPLC-Separation of Heteronuclear Oligometallocenes. Isolation of Cations of Rhodocenylferrocene, 1,1'-Dirhodocenylferrocene, and 1-Cobaltocenyl-1'-rhodocenylferrocene », Chromatographia, vol. 30, nos 9–10, , p. 543–545 (DOI 10.1007/BF02269802)
- P. Jaitner, H. Schottenberger, S. Gamper et D. Obendorf, « Termetallocenes », J. Organomet. Chem., vol. 475, nos 1–2, , p. 113–120 (DOI 10.1016/0022-328X(94)84013-X)
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Rhodocene » (voir la liste des auteurs).
 Portail de la chimie
Portail de la chimie