Lymphome de Hodgkin
Le lymphome de Hodgkin (LH) ou lymphome hodgkinien (par opposition au lymphome non hodgkinien) est un type de lymphome (cancer du système lymphatique) caractérisé par la présence de grandes cellules atypiques, les cellules de Reed-Sternberg. Le fait qu'il s'agisse du premier lymphome bien caractérisé a conduit à appeler lymphomes non hodgkiniens (LNH), par exclusion, tous les autres types de lymphome.
Ne doit pas être confondu avec Lymphome non hodgkinien.
Pour les articles homonymes, voir Hodgkin.

| Spécialité | Oncologie |
|---|
| CISP-2 | B72 |
|---|---|
| CIM-10 | C81 |
| CIM-9 | 201 |
| ICD-O | 9650/3-9667/3 |
| OMIM | 300221 et 400021 236000, 300221 et 400021 |
| DiseasesDB | 5973 |
| MedlinePlus | 000580 |
| eMedicine | 201886 |
| eMedicine | med/1022 |
| MeSH | D006689 |
| Causes | Infection à virus d'Epstein Barr (en) |
| Médicament | Lomustine (en) et chlorambucil |
| Patient UK | Hodgkins-lymphoma-pro |
![]() Mise en garde médicale
Mise en garde médicale
La cellule de Reed-Sternberg est indispensable au diagnostic, mais elle n'est pas totalement spécifique, et peut se retrouver (rarement) dans d'autres types de lymphomes (lymphomes T périphériques en particulier). Sa nature a été longtemps débattue mais il est maintenant bien établi qu'il s'agit d'une cellule de la lignée lymphoïde B[1].
Ses synonymes incluent « maladie de Hodgkin-Paltauf-Sternberg », « Hodgkin », « lymphome hodgkinien », « granulomatose maligne », « lymphogranulomatose maligne » et « cancer des ganglions ».
Physiopathologie
L'origine des cellules caractéristiques du lymphome de Hodgkin, les cellules de Hodgkin (mononucléées) et les cellules de Reed-Sternberg (multinucléées), a été pendant très longtemps débattue et ce n'est qu'au tournant de ce siècle qu'il a été formellement établi que ces cellules étaient d'origine lymphoïde B[1]. Cette identification tardive, plus de 150 ans après la description de la maladie, est due au fait que ces cellules tumorales ne représentent le plus souvent que 1 % de la masse tumorale, majoritairement constituée d'un infiltrat cellulaire réactionnel. Par ailleurs ces cellules n'expriment pas de marqueurs typiques d'une origine lymphoïde B ou T.
La démonstration que toutes les cellules tumorales portent des réarrangements des gènes des immunoglobulines avec présence par ailleurs des mutations somatiques caractéristiques de la lymphogenèse B traduit le fait que ces cellules sont originaires des centres germinatifs du ganglion. Dans une proportion notable de cas, ces mutations somatiques sont caractérisées comme défavorables, ce qui aurait dû conduire à l'élimination par apoptose de ces cellules ; cette non-élimination est à l'origine, dans certains cas, de la transformation maligne de ces lymphocytes B[1].
Enfin, de nombreuses études récentes ont pu démontrer que les cellules tumorales étaient caractérisées par une modification très importante du profil normal d'expression génique : la plupart des gènes normalement exprimés dans les lymphocytes B ne le sont plus, alors que de nombreuses voies de transduction ou de transcription sont dérégulées[1].
Signes cliniques
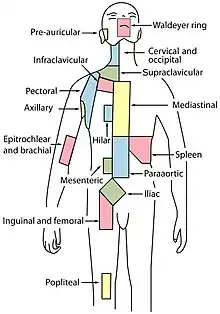
L'augmentation de taille des ganglions lymphatiques (adénopathies) est le signe le plus fréquent ; les adénopathies sont fermes, mais indolores (en dehors des classiques mais rares douleurs à l'ingestion d'alcool) et non inflammatoires. Elles sont localisées le plus souvent au niveau des aires ganglionnaires cervicales, sus-claviculaires ou axillaires. L'examen peut aussi retrouver des adénopathies des aires inguinales ou crurales, une splénomégalie ou une hépatomégalie.
Certains symptômes peuvent être secondaires à la présence de ganglions profonds, en particulier des signes pulmonaires : toux chronique et sèche voire dyspnée, et des signes cardiaques : syndrome cave supérieur, en cas d'adénopathies médiastinales compressives (situation rare dans le lymphome de Hodgkin).
Environ un tiers des malades présentent aussi des signes généraux (dits signes ou symptômes « B ») : amaigrissement (significatif s'il est supérieur à 10 % du poids du corps), asthénie, fièvre (significative si > 38° pendant au moins 7 jours), ou sueurs nocturnes et abondantes (significatives si elles imposent de changer son linge). Un prurit isolé et intense est un signe classique et précède parfois de beaucoup la date du diagnostic.
Diagnostic


Le diagnostic de lymphome de Hodgkin peut être évoqué sur une ponction d'un ganglion. L'analyse d'un frottis de suc ganglionnaire peut en effet révéler la présence de cellules de Sternberg.
Mais le diagnostic formel de lymphome de Hodgkin repose sur l'étude anatomopathologique d'un ganglion. Ceci impose une biopsie d'une adénopathie réalisée soit de façon chirurgicale (exérèse d'un ganglion), soit par ponction-biopsie au trocart d'un ganglion, faite par voie externe, par un radiologue, sous contrôle d'une échographie ou d'un scanner.
L'examen anatomopathologique révèlera l'association de la présence de grandes cellules malignes (dites de Reed-Sternberg [RS]), binucléées, avec des nucléoles proéminents, et d'une destruction de l'architecture normale du ganglion. Il existe également une importante réaction cellulaire faite de lymphocytes T, d'histiocytes et d'éosinophiles. À cette étude anatomopathologique, sera couplée une étude de l'immunophénotype des cellules tumorales. Les cellules sont typiquement CD15+ et CD30+, souvent CD25+ et, dans près de la moitié des cas LMP+ (marqueur traduisant que les cellules de Reed-Sternberg contiennent le génome du virus d'Epstein-Barr) ; le CD20 (marqueur B) est le plus souvent négatif; sa positivité devrait faire évoquer un lymphome de Popemma, doit être en revanche vérifiée.
L'anatomopathologiste classera également le lymphome de Hodgkin en un de ses sous-types histologiques (cf. infra) en sachant que cette classification ne modifie pas, le plus souvent, le traitement qui sera administré.
Sous-types histologiques
Le lymphome de Hodgkin peut être classé en quatre sous-types histologiques en fonction des données de l'examen anatomopathologique.
Le classement établi par Rye en 1965 distingue quatre types[2] :
- le type 1 riche en lymphocytes ou à prédominance lymphocytaire (PL) ;
- le type 2 scléronodulaire (SN) associé à la présence de sclérose ;
- le type 3 à cellularité mixte (CM) ;
- le type 4 à déplétion lymphocytaire (DL).
Plus récemment, la classification OMS a exclu de ce cadre les formes nodulaires riches en lymphocytes, considérées maintenant comme une entité à part (NLPH des anglo-saxons, lymphome de Popemma, paragranulome de Popemma et Lennert). Selon cette classification[3] il est désormais question de lymphome de Hodgkin classique avec comme sous-entités :
- la forme avec sclérose nodulaire ;
- la forme riche en lymphocytes ;
- la forme avec cellularité mixte ;
- la forme avec déplétion lymphoïde.
Enfin, la forme avec sclérose nodulaire est subdivisée en deux sous-groupes en fonction de la rareté (NS1) ou de la richesse (NS2) en cellules de Sternberg.
Bilan d'extension
Le diagnostic posé avec certitude, l'hématologue doit effectuer un bilan d'extension de la maladie. Ce bilan nécessite :
- un examen clinique complet ;
- des examens d'imagerie : radiographie de thorax, échographie abdominale, scanner (tomodensitométrie : TDM) cervical, thoracique et abdominopelvien. La lymphographie n'est plus pratiquée. Une tomographie par émission de positons (TEP) au 18F-FDG est maintenant systématique ; extrêmement sensible, elle permet de détecter des atteintes non vues par les examens d'imagerie classique et évite ainsi de sous-stader des patients. La TEP a remplacé la scintigraphie au Gallium qui n'est plus réalisée. Une échographie cardiaque peut être nécessaire pour préciser l'atteinte ou non du péricarde ;
- un bilan sanguin comportant un dosage des LDH, un hémogramme, un bilan hépatique, un bilan martial, une électrophorèse des protides et un bilan inflammatoire ; il permet de réunir les éléments de l'index pronostique (cf. infra) ;
- une biopsie ostéomédullaire (BOM) est surtout indiquée dans les formes avec signes B ou les formes étendues ; l'intérêt de cette BOM est actuellement rediscuté car la TEP permet d'identifier le plus souvent les atteintes ostéomédullaires ;
- plus rarement, d'autres examens pourront être nécessaires afin de préciser l'extension de la maladie : ponction-biopsie du foie (PBH), ponction ou biopsie pleurale, etc. Ces examens, « agressifs » seront surtout nécessaires dans les cas où leur positivité modifierait le stade de la maladie.
Autres examens initiaux
Un certain nombre d'autres examens seront réalisés au diagnostic, essentiellement pour être sûr qu'il n'y a pas une atteinte d'organe antérieure ou une contre-indication à l'utilisation de certains médicaments. Un électrocardiogramme ainsi qu'une échographie cardiaque par voie transthoracique sont réalisés avant toute chimiothérapie par anthracycline. Des épreuves fonctionnelles respiratoires sont réalisées avant une chimiothérapie par bléomycine.
Une congélation de sperme dans un Centre d'étude et de conservation des œufs et du sperme humains est systématiquement proposée aux adolescents et aux hommes adultes car certaines chimiothérapies utilisées dans le lymphome de Hodgkin sont stérilisantes pour les patients de sexe masculin.
Stades
Au terme du bilan d'extension, le stade de la maladie selon la classification d'Ann Arbor sera déterminé ; celle-ci distingue :
- le stade I : une seule aire ganglionnaire est atteinte ;
- le stade II : au moins deux aires ganglionnaires, d'un seul côté du diaphragme, sont atteintes ;
- le stade III : atteinte d'aires ganglionnaires de part et d'autre du diaphragme ;
- le stade IV : atteinte d'un ou de plusieurs organes extralymphatiques (poumon, foie, os, moelle osseuse, etc.).
NB1 : la rate compte comme une aire ganglionnaire ; l'envahissement de la rate peut être noté par un « s » ; une atteinte hépatique en revanche définit un stade IV de même que l'atteinte d'autres organes.
NB2 : L'envahissement d'un organe extralymphatique ou des séreuses (plèvre, péricarde) par contiguïté à partir d'un ganglion envahi (par exemple : envahissement du parenchyme pulmonaire ou de la paroi thoracique antérieure à partir d'une adénopathie médiastinale) est noté par un « E ».
L'absence ou la présence de signes généraux est notifiée en rajoutant un « A » ou un « B ». Les cas en « a » ou « b » peuvent être classés en fonction de la présence ou non de signes biologiques d'inflammation (cette classification n'est plus guère utilisée actuellement).
Sont distinguées ainsi les formes dites localisées (Stades I et II) et les formes étendues (Stades III et IV) qui relèvent de traitements d'intensité différente. Les stades possibles vont ainsi de IA à IVBE. À noter que la présence de signes B, ou d'une extension par contiguïté, peut à elle seule imposer un changement de groupe thérapeutique. Une forme IIBE sera ainsi de pronostic plus sévère qu'une forme IIIA.
Facteurs pronostics
Le facteur pronostique principal est le degré d'extension de la maladie. D'autres facteurs pronostiques sont également considérés et peuvent être utilisés pour la statification des patients :
- l'importance du syndrome tumoral (caractère bulky des anglo-saxons) : certains protocoles prennent en compte la présence ou non de très grosses adénopathies (> 6 cm ou > 10 cm) ou la présence ou non d'un gros médiastin (défini par un rapport médiastin/thorax > 0,33 ou > 0,45) ;
- l'existence ou non d'une extension par contiguïté ;
- une étude internationale a identifié en 1998 sept facteurs ayant une valeur pronostique pour les stades avancés[4] :
- sexe masculin,
- âge ≥ 45 ans,
- stade IV de la maladie (versus stade III),
- hémoglobine < 10,5 g/dL,
- nombre de lymphocytes < 600/μl ou < 8 %,
- nombre de globules blancs ≥ 15 000/μl,
- albumine < 4,0 g/dl.
Dans cette étude un patient n'ayant aucun de ces facteurs a une chance de survie à 5 ans de 84 %. Le pronostic décroit ensuite en fonction du nombre de facteurs présents, la survie à 5 ans tombant à 42 % pour les patients ayant 5 de ces facteurs.
Le caractère inflammatoire est parfois plus simplement pris en compte par le degré d'augmentation de la vitesse de sédimentation à la 1re heure (> 30 mm ou > 50 mm).
Le type histologique « sclérose nodulaire » est considéré comme de pronostic plus sévère et peut intervenir dans la stratification également.
Enfin la réponse tumorale est également prise en compte. Elle est évaluée de deux façons : la diminution volumétrique de la tumeur (les mensurations des masses tumorales sont quantifiées par un TDM) et la persistance ou non d'une fixation positive au TEP. Cette évaluation peut se faire de façon précoce (après seulement 2 cures) ou en fin de traitement. Selon la qualité de la réponse observée, il sera décidé, selon les protocoles, d'alléger ou d'intensifier le traitement. Par exemple, beaucoup de protocoles actuels proposent de ne pas irradier les patients qui ont une très bonne réponse à la chimiothérapie.
Traitement
Avec un traitement approprié, plus de 90 % des maladies de Hodgkin sont curables[réf. nécessaire]. Le traitement associe classiquement une chimiothérapie et/ou une radiothérapie en sachant que cette dernière est de moins en moins utilisée. Le choix thérapeutique s'effectue lors d'une réunion de concertation pluridisciplinaire (comme pour tout cancer). Les traitements étant en cours d'évolution, il peut être proposé au patient d'intégrer un protocole thérapeutique (cf. : thérapies ciblées).
Chimiothérapie
L'intensité de la chimiothérapie va dépendre du degré d'extension de la maladie. Selon la stratification choisie par le protocole, les patients sont répartis en groupes thérapeutiques en fonction du stade et de la présence ou non de certains facteurs pronostiques.
La chimiothérapie anticancéreuse la plus utilisée pour les patients adultes est l'ABVD qui associe l'Adriamycine, la Bléomycine, la Vinblastine et la Dacarbazine. Mise au point en Italie dans les années 1970, l'ABVD reste le gold-standard auquel les autres types de cure doivent se comparer. Les cures d'ABVD sont répétées tous les 28 jours pour un total de 3 à 8 cures selon le degré d'extension et les protocoles.
D'autres types de cures sont utilisés :
- les cures de type BEACOPP (mises au point par le groupe allemand), plus intensives, sont utilisées surtout par les groupes européens. Le BEACOPP dit escaladé est encore plus lourd ;
- en pédiatrie, l'utilisation des anthracyclines sera restreinte ; les cures de type OEPA, COPP, COPDAC, etc. sont utilisées ;
- les classiques cures de MOPP (méthylchlorétamine, vincristine, procarbazine et prednisone) ne sont plus utilisées, au moins en première ligne.
En cas de rechute, des cures de chimiothérapie apportant des agents non utilisés en première ligne sont utilisées. Les principales cures sont l'IVA, l'ICE, le DHAP, le MINE, etc.
Radiothérapie


Le lymphome de Hodgkin est une maladie très radiosensible (c'est-à-dire qu'il « répond » bien à la radiothérapie). Malheureusement la radiothérapie est aussi la principale source de séquelles secondaires au traitement, raison pour laquelle la médecine cherche à réduire ses indications et son intensité.
Un des moyens de réduire l'intensité de la radiothérapie est de l'associer à une chimiothérapie. Ainsi, l'attitude de choix est le plus souvent un traitement dit combiné[5].
La dose de radiothérapie délivrée était classiquement importante : 36 voire 40 Gray ; progressivement celle-ci est réduite à 25 voire 20 Gy.
Les champs d'irradiation se sont également réduits au cours du temps : d'une irradiation lymphoïde totale ou d'une irradiation étendue (classique mantelet par exemple), le radiothérapeute est passé à une irradiation dite en involved-field ou IFRT (seule l'aire atteinte est irradiée), voire à une irradiation « nodulaire » ou INRT (seul le ganglion atteint est irradié).
Actuellement, il est considéré qu'une forme localisée ne doit pas être traitée par une radiothérapie exclusive (le traitement combiné permet de délivrer une irradiation moins intensive) ; que les formes étendues, stade IV en particulier, doivent être traitées le plus souvent, au moins chez l'adulte, par une chimiothérapie exclusive (afin d'éviter une irradiation étendue) ; et que les patients bon répondeurs précoces à la chimiothérapie peuvent sans doute ne pas être irradiés (évaluation en cours).
L'irradiation se fait en plusieurs séances (il est question d'irradiation fractionnée).
La zone sus-diaphragmatique (mantelet, etc.) doit être irradiée, puis si besoin la zone sous-diaphragmatique (Y inversé, barre lombosplénique, etc.). Une irradiation sus ou sous diaphragmatique s'étale en général sur 2 à 3 semaines en fonction de la dose totale délivrée.
Les principaux effets secondaires de la radiothérapie sont : le défaut de croissance de la zone irradiée chez l'enfant, les atteintes thyroïdiennes (hypothyroïdie, cancer), l'insuffisance ovarienne (ménopause précoce voire stérilité quand les ovaires sont dans le champ d'irradiation et n'ont pas été déplacés avant celle-ci), les atteintes coronaires : sténose des artères (angor, infarctus du myocarde), etc. mais aussi les tumeurs malignes (cancer du sein, ostéosarcome, etc.) et les leucémies aiguës myéloblastiques ou les myélodysplasies (le risque d'hémopathie maligne secondaire est aussi augmenté par certaines chimiothérapies, alkylants en particulier).
La fréquence attendue de ces complications dans les protocoles actuels sera, a priori, très inférieure à celle observée dans les protocoles anciens compte tenu des modifications de la prise en charge faites au cours du temps.
Greffes de moelle
L'autogreffe est effectuée à partir d'un prélèvement de moelle autologue, ou plus souvent ici à partir d'un prélèvement de cellules souches hématopoïétiques (CSH) circulantes obtenu par cytaphérèse après mobilisation par une cure de chimiothérapie adaptée et un facteur de croissance de la lignée granuleuse (G-CSF). Il s'agit ici de jouer sur un « effet dose » : une chimiothérapie très intensive, associée ou non à une irradiation corporelle totale, est administrée au patient et les CSH sont réinjectées ensuite, ce qui permettra au patient de sortir d'aplasie dans un délai moyen de 3 semaines après la réinjection.
L'autogreffe est indiquée en première ligne pour des patients réfractaires ou ayant des critères pronostiques initiaux très péjoratifs et en cas de rechute. Certains protocoles utilisent une approche de double autogreffe.
L'allogreffe de moelle reste en partie expérimentale dans le traitement du lymphome de Hodgkin mais un effet greffe anti-lymphome de Hodgkin est bien établi : le risque de rechute est diminué (par comparaison avec une autogreffe) grâce à un effet allogénique : élimination d'éventuelles cellules tumorales résiduelles par les cellules immunitaires du donneur. L'allogreffe est indiquée surtout chez des patients en rechute après une autogreffe. Les modalités de réalisation sont les mêmes qu'une autogreffe : conditionnement (chimiothérapie intensive associée ou non à une irradiation corporelle totale) puis réinjection des CSH. Pour certains patients fragiles le conditionnement à la greffe sera atténué (on parle alors de « mini-greffe »). Les suites d'une allogreffe sont plus longues et potentiellement plus compliquées que celles d'une autogreffe. La prévention de la maladie du greffon contre l'hôte impose en particulier une immunosuppression prolongée et celle-ci, de même qu'une maladie du greffon contre l'hôte mal contrôlée, alourdit beaucoup les suites de la greffe. Le contrôle de la maladie du greffon est majeur pour la réussite de l'allogreffe : elle est souhaitable car sa survenue est associée à une diminution du risque de rechute, mais elle ne doit pas être trop toxique pour le patient. Enfin la mortalité post allogreffe est supérieure à celle de l'autogreffe. Tout ceci explique pourquoi l'allogreffe est généralement proposée à des cas plus sévères.
Thérapies ciblées
De nombreux nouveaux médicaments sont actuellement en cours d'étude (phases II et III) dans le lymphome de Hodgkin : il s'agit d'anticorps monoclonaux (anti-CD30) ou de médicaments inhibiteurs des histones déacétylases (panobinostat) ou de m-TOR[6]. Le SGN-35 ou brentuximab védotine, anti-CD30 couplé à une chimiothérapie, l'aurostatine, apparait ainsi très prometteur[7].
Enfin, certains évaluent l'intérêt d'un monoclonal anti-CD20 (le rituximab) ; bien que la cellule de Sternberg n'exprime pas le CD20, une théorie actuelle fait l'hypothèse que les cellules souches tumorales, elles, seraient CD20+ et que l'élimination de ces cellules souches permettraient de supprimer le risque de rechute.
La place de ces nouveaux agents dans le traitement du lymphome de Hodgkin reste néanmoins à définir.
Traitements symptomatiques
Ces traitements visent notamment à prévenir certaines complications de la chimiothérapie. Ainsi, de l'érythropoïétine (EPO) pourra être prescrite aux patients anémiques, tout comme des facteurs de croissance granulocytaires (G-CSF) aux patients aplasiques. Des traitements devront également prévenir les infections opportunistes, les vomissements ainsi que la douleur.
Épidémiologie
Épidémiologie descriptive
Contrairement aux autres lymphomes, dont la fréquence augmente avec l'âge, les lymphomes hodgkiniens ont une courbe de fréquence bimodale : en effet, ils interviennent plus fréquemment au sein de deux groupes d'âge distincts, le premier groupe d'âge étant celui des jeunes adultes entre 20 et 30 ans et le second vers 70 ans. Cette maladie touche plus fréquemment les hommes, sauf dans le cas des formes avec sclérose nodulaire, sous-type histologique qui touche plus fréquemment les femmes. Selon une étude récente de la revue médicale Lancet, son incidence, d'environ 4 cas sur 100 000, augmenterait chez les jeunes adultes. Cette maladie représente toutefois moins de 1 % de la totalité des cancers connus.
En France, le rapport de l'Institut du Cancer publié en 2009, estime le nombre de cas pour l'année 2005 à 1544 (757 femmes, 787 hommes) ; ceci en fait une cause rare de cancer : 0,5 % des cancers et 24e rang sur les 25 localisations étudiées. Depuis 1980, l'incidence annuelle diminue chez les hommes alors qu'elle est en augmentation chez les femmes : +3,3 % par an de 2000 à 2005[8].
La maladie est encore plus rare chez l'enfant et concerne essentiellement des enfants de plus de 10 ans et des adolescents. Parmi les très rares petits enfants atteints il y a une très forte prédominance de garçons. Le nombre de cas estimé d'âge pédiatrique est d'une centaine par an en France.
Épidémiologie analytique
L'étiologie du lymphome de Hodgkin n'est pas élucidée. Certains facteurs qui interviennent dans l'oncogénèse de ce lymphome ont été néanmoins identifiés.
Le virus d'Epstein-Barr - responsable de la mononucléose infectieuse - semble jouer un rôle dans certaines formes de lymphome de Hodgkin. Le virus EBV est présent dans les cellules de Reed-Sternberg (RS) dans 40 % des cas de LH classiques des patients des pays développés, particulièrement dans les formes à cellularité mixte et celles avec déplétion en lymphocytes ; cette proportion est plus importante chez l'enfant et dans les pays émergents : 90 % des cas par exemple pour les cas pédiatriques d'Amérique centrale ou d'Amérique du Sud[1].
Les patients infectés par le VIH ont également une incidence augmentée de lymphomes, dont le lymphome de Hodgkin ; chez eux, l'association avec l'EBV est quasi constante[1].
Dans les cellules RS sont exprimées trois protéines virales (EBNA1, LMP1 et LMPA2) et aussi deux ARN non codants. Il est établi que ces protéines virales jouent un rôle dans le processus tumoral ; dans les cas où les cellules RS ne contiennent pas le génome de l'EBV, des mutations d'un gène, TNFAIP3 pourraient « remplacer » le virus[1].
L'exposition de la mère à des pesticides domestiques (les insecticides semblent en cause) durant sa grossesse serait un facteur de risque pour l'enfant (risque doublé), sauf pour le lymphome hodgkinien de type sclérose nodulaire (nodular sclerosis ou NSHL pour les anglo-saxons), forme qui touche surtout des filles plus âgées[9].
Histoire
Il a été décrit pour la première fois par Sir Thomas Hodgkin en 1832[10]. Un demi-siècle plus tard, Wilms propose l'appellation maladie de Hodgkin[11]. En 1902, Dorothy Reed[12] et Carl Sternberg[13] décrivent les cellules caractéristiques du lymphome de Hodgkin : les cellules de Hodgkin, mononucléées, et les cellules de Reed-Sternberg, polynucléées. L'origine du lymphome de Hodgkin a été longtemps discutée : inflammatoire, infectieuse (tuberculose en particulier) ou tumorale.
La première tentative de chimiothérapie date de 1947 avec un dérivé du gaz moutarde[14] avec un succès très relatif. Le pronostic en était effroyable puisque la durée de vie ne dépassait pas deux ans après le diagnostic[15]. Des progrès très significatifs ont été atteints avec la mise au point, pour la première fois en médecine, de polychimiothérapies. La première cure a été le MOPP en 1964[16] (mechlorethamine, vincristine, procarbazine et prednisone) puis l'ABVD (doxorubicine, bléomycine, vinblastine et dacarbazine) dans le milieu des années 1970[17].
Patients célèbres
- Probablement, la romancière Jane Austen en 1817[18]
- Howard Carter, archéologue et égyptologue, mort en 1939.
- Le pianiste roumain Dinu Lipatti, décédé en 1950
- Le joueur du hockey canadien Mario Lemieux en 1993[19],[20]
- Le réalisateur-scénariste et acteur Nanni Moretti parle de sa guérison dans son film Journal intime en 1993[21]
- L'acteur Richard Harris en est mort en 2002[22]
- L'acteur Michael C. Hall en a souffert en 2010[23]
- L'auteur-compositeur et chanteur Éric Charden en est mort le 29 avril 2012[24],[25]
- La joueuse de tennis Victoria Duval déclare sa maladie en 2014.
- Le planchiste québécois médaillé olympique Maxence Parrot a reçu un diagnostic en [26]
- La tenniswoman Carla Suárez Navarro déclare la maladie publiquement en septembre 2020[27].
Dans la culture
- 1994 : Journal intime (Caro Diario) : le personnage principal du film est atteint de cette maladie.
- 2008 : Comme une étoile dans la nuit : le personnage principal du film est atteint de cette maladie.
Notes et références
- (en) Küppers R.The biology of Hodgkin's lymphoma.Nat Rev Cancer. 2009 Jan;9(1):15-27
- « La maladie de Hodgkin », www.leucemie-espoir.org (consulté le )
- Jaffe ES.The 2008 WHO classification of lymphomas: implications for clinical practice and translational research. Hematology Am Soc Hematol Educ Program. 2009:523-31.
- Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin's disease. International Prognostic Factors Project on Advanced Hodgkin's Disease.N Engl J Med. 1998 Nov 19;339(21):1506-14.
- « Lymphomes malins - Les principes thérapeutiques », www.uvp5.univ-paris5.fr, (consulté le )
- Proceedings of the 8th International Symposium on Hodgkin Lymphoma. Cologne, Germany, October 23-26, 2010. Haematologica, 95 (S4), p.S1-S152
- Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL, Forero-Torres A.Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas.N Engl J Med. 2010 Nov 4;363(19):1812-21
- <La situation du Cancer en France en 2009. Institut National du Cancer. http://www.e-cancer.fr/component/docman/doc_download/1285-la-situation-du-cancer-en-france-en-2009
- Household Exposure to Pesticides and Risk of Childhood Hematopoietic Malignancies: The ESCALE Study (SFCE) Environ Health Perspect 115:1787-1793 (2007). doi: 10.1289/ehp.10596], Jérémie Rudant et al. décembre 2007, Environmental Health Perspectives, Vol. 115 | N° 12, en ligne depuis le 25 septembre 2007
- Hodgkin T, On some morbid appearances of the absorbent glands and spleen, Med Chir Trans, 1832;17:69-97
- Wilks S, Cases of enlargement of the lymphatic glands and spleen (or Hodgkin's disease), with remarks, Guys Hosp Rep 1865;11:56-67
- Reed D. On the pathological changes in Hodgkin's diseasewith special reference to its relation to tuberculosis. Johns Hopkins Hosp Rep, 1902, 10: 133-193
- Sternberg C. Übe r eine eigenartige unter dem Bilde der Pseudoleukämie verlaufende Tuberkulose des lymphatischen Apparates. Z. Heilkunde, 1898, 19: 21-90
- Goodman LS, Wintrobe MM, Dameshek W et Als. Nitrogen mustard therapy: use of methyl-bis(beta-chloroethyl)amine hydrochloride and tris(beta-chloroethyl)amine hydrochloride for Hodgkin's disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders, JAMA, 1946;132:126-132
- Diehl V, Hodgkin's Disease — From Pathology Specimen to Cure, N Eng J Med, 2007;357:1968-1971
- Devita VT Jr, Serpick AA, Carbone PP, Combination chemotherapy in the treatment of advanced Hodgkin's disease, Ann Intern Med, 1970;73:881-895
- Bonadonna G, Zucali R, Monfardini S et Als. Combination chemotherapy of Hodgkin's disease with adriamycin, bleomycin, vinblastine, and imidazole carboxamide versus MOPP, Cancer, 1975;36:252-259
- Note : voir la page Causes de la mort de Jane Austen
- (en) « Lemieux Has Hodgkin’s Disease; Doesn’t Appear Life-Threatening », Los Angeles Times, (lire en ligne)
- (en) « Hockey; Lemieux Stricken By Hogdkin's Disease », The New York Times, (lire en ligne)
- Carmela Lettieri, « Le symbolisme des étapes dans Journal intime de Nanni Moretti », Cahiers d'études romanes, no 17, , p. 229-241 (lire en ligne)
- Actor Richard Harris dies, BBC News, 25 octobre 2002
- Los Angeles Times, 13 janvier 2010
- Interview par Caroline Rochmann, « Eric Charden. “Gabrielle, mon barrage contre la maladie” », sur Paris Match,
- Stone a perdu Charden, Le Point, le 29 avril 2012.
- Tommy Thurber, « Le planchiste québécois Maxence Parrot frappé par le cancer » (consulté le )
- « Carla Suarez Navarro souffre d'un lymphome », L'Equipe, (lire en ligne)
Voir aussi
Bibliographie
Ségolène de Margerie, Encore combien de jours, Maman ?, témoignage.
Filmographie
- Journal intime de Nanni Moretti. Chapitre 3 : « Les médecins ».
- Sweet November (2001) de Pat O'Connor.
- Comme une étoile dans la nuit (2008) de René Féret.
Articles connexes
Liens externes
- Notices dans des dictionnaires ou encyclopédies généralistes :
- Ressources relatives à la santé :
- Classification internationale des maladies oncologiques
- NCI Thesaurus
- Orphanet
- (en) Classification internationale des soins primaires
- (en) Diseases Ontology
- (en) DiseasesDB
- (en + es) Genetic and Rare Diseases Information Center
- (en) Héritage mendélien chez l'humain
- (en) Héritage mendélien chez l'humain
- (en) Medical Subject Headings
- (en + es) MedlinePlus
- (cs + sk) WikiSkripta
- Le lymphome sur le site du Groupe d'étude du lymphome de l'adulte
- (en) Hodgkin's Lymphoma Home Page sur le site de l'institut national américain sur le cancer.
- (en) Historique de la maladie d'Hodgkin
 Portail de la médecine
Portail de la médecine  Portail de l’hématologie
Portail de l’hématologie