Syndrome de Guillain-Barré
Le syndrome de Guillain-Barré (SGB) ou de Guillain-Barré-Strohl est une maladie auto-immune inflammatoire du système nerveux périphérique. Elle se manifeste brutalement par une parésie, qui prédomine en proximal des muscles. Elle s'accompagne d'une absence ou réduction des réflexes (paralysie flasque), et de troubles sensitifs. Dans la plupart des cas, les personnes atteintes récupèrent en quelques mois, mais il existe des formes graves. Avec la quasi-disparition de la poliomyélite, le SGB est devenu la première cause de paralysie flasque aiguë dans le monde. De nos jours, il est considéré comme une des principales urgences en neurologie. Le SGB est aussi connu sous les noms suivants :
- Polyneuropathie aiguë inflammatoire démyélinisante
- Polyradiculonévrite aiguë idiopathique
- Polynévrite aiguë idiopathique
- Paralysie ascendante de Landry
| Spécialité | Neurologie |
|---|---|
| Symptôme | Paralysie ascendante (d) |
| CISP-2 | N94 |
|---|---|
| CIM-10 | G61.0 |
| CIM-9 | 357.0 |
| OMIM | 139393 |
| DiseasesDB | 5465 |
| MedlinePlus | 000684 |
| eMedicine | 792008, 1169959, 315632 et 1180594 |
| eMedicine | emerg/222 neuro/7pmr/48neuro/598 |
| MeSH | D020275 |
![]()
Historique
Au moins deux descriptions du XIXe siècle pourraient correspondre à des SGB. En 1828, Chomel décrit une épidémie de polynévrites aiguës[1]. En 1859, Octave Landry, décrit une maladie grave paralysant les jambes, les bras, le cou et les muscles respiratoires (paralysie ascendante de Landry, maladie ou syndrome de Landry)[2]. Des observations du même genre se retrouvent dans d'autres pays. En 1891, Quincke découvre la possibilité de prélever et d'analyser le liquide céphalo-rachidien par ponction lombaire. C'est la première approche biologique directe de ces maladies neurologiques.
En 1916, Georges Guillain, Jean Alexandre Barré et André Strohl présentent deux cas de paralysie généralisée transitoire avec une anomalie caractéristique du liquide céphalo-rachidien : élévation anormale des protéines, et un compte normal de cellules (dissociation albumino-cytologique)[3]. Dans les années 1930-1950, la recherche d'un virus causal spécifique ne donne rien. Dans les années 1960, la nature immunologique de la maladie est affirmée. Dans les années 1980, premières identifications des anticorps liés à la maladie[4].
À l'origine, et jusqu'à la fin des années 1930, Guillain et Barré ont insisté sur l'évolution favorable de l'affection, pour en faire le deuxième critère déterminant du diagnostic[5], d'où une réputation historique de bénignité qui, à tort, peut encore persister aujourd'hui [6]. Depuis le milieu du XXe siècle, l'évolution favorable n'est plus un critère de diagnostic. Après 1976, à la suite d'une série de cas liés à une vaccination contre une menace de grippe porcine aux États-Unis, les critères de diagnostic font l'objet d'une normalisation internationale[7], périodiquement réévaluée. Ce qui permet, sous le terme simplifié de " Syndrome de Guillain - Barré ", de regrouper de nombreuses formes de paralysies extensives aiguës, anciennement ou nouvellement décrites, y compris les formes graves.
Épidémiologie
La fréquence de la maladie est d'environ 1,5 sur 100 000 personnes par an (entre 0,8 et 1,9 dans les pays occidentaux). Rare avant l'âge de cinq ans, le risque s'accroît avec l'âge et touche 3 hommes pour 2 femmes[8]. Les formes familiales sont exceptionnelles[9]. La récidive est également rarissime.
L'origine du SGB n'est pas connue, mais il existe des facteurs déclenchants retrouvés de 1 à 6 semaines avant le début de la maladie. Il s'agit le plus souvent d'une infection (surtout digestive ou respiratoire), parfois d'une vaccination, d'un acte chirurgical, ou d'un évènement stressant[10]. Il peut survenir aussi au cours de maladies systémiques (lupus érythémateux) ou de la maladie de Hodgkin.
Le syndrome de Guillain-Barré peut également survenir durant la grossesse. Heureusement, des épisodes répétés ne se représentent habituellement pas lors de grossesses ultérieures.
Facteurs infectieux
Dans 2/3 des cas, la maladie survient 1 à 3 semaines après une infection bactérienne à Campylobacter jejuni (gastro-entérite et diarrhées) ou virale à Cytomégalovirus (fièvre). Le SGB survient 1 à 2 fois sur 1000 infections à cytomégalovirus, et un peu moins de 1 fois sur 1000 infections à Campylobacter. Cette rareté pourrait s'expliquer par une susceptibilité particulière de certains individus, mais la prédisposition génétique unique est peu probable, car le SGB ne se manifeste pas au sein d'une même famille. En revanche, des petites épidémies ont été notées en Chine lors de gastro-entérites liées à la contamination de l'eau par Campylobacter jejuni[11].
Les autres infections en cause sont celles à VIH, Epstein-Barr, Mycoplasma pneumonia, Haemophilus influenza, varicelle, etc[10]. L'augmentation des SGB au cours des épidémies à virus zika serait liée à une co-infection avec d'autres arboviroses (dengue, chikungunya...) circulant dans la même région[12].
Grippe et vaccins
L’incidence du développement d'un SGB serait de 4 à 7 cas pour 100.000 sujets grippés[13]. En 1976, un risque accru de SGB est relevé après une vaccination antigrippale.
La fausse alerte pandémique de 1976
Début janvier 1976, aux États-Unis, une épidémie de grippe débute sur la base militaire de Fort Dix, dans le New-Jersey, entrainant le décès d'au moins un soldat. En février, la souche virale est isolée, c'est un virus de grippe porcine H1N1 ressemblant à celui de la pandémie de 1918-1919. L'affaire est porté au plus haut niveau de l'administration américaine. Un comité d'experts, désignés à cette occasion, estime qu'une grave pandémie se prépare et suggère de produire un vaccin pour une campagne de masse dès l'automne suivant. Fin mars, le Président Gerald Ford annonce à la télévision qu'il dégage 135 millions de dollars pour un programme de vaccinations[14].
Pendant l'été 1976, 7000 personnes sont vaccinées sans problème, ce qui permet de lancer la campagne dès la rentrée. Fin novembre, les premiers cas de SGB, survenant 2 à 3 semaines après vaccination, sont notifiés. A la mi-décembre, l'enquête épidémiologique révèle un risque accru de SGB (de 7 à 8 fois). La pandémie attendue n'ayant pas lieu, la campagne (près de 45 millions de vaccinés) est immédiatement stoppée[15]. Depuis de nombreuses études ultérieures ont confirmé la relation de causalité[16],[17].
La surveillance ultérieure
L'événement a suscité une surveillance renforcée et continue, jusqu'à nos jours, des autres vaccins antigrippaux. Durant la période 1976-2008, on trouve un léger risque accru de 1,3 à 2 cas supplémentaires pour 1 million de personnes. Lors de la vaccination antigrippale contre le H1N1 de 2009-2010 , ce risque est de 1 à 6 cas par million de personnes vaccinées aux États-Unis[18], 1 à 3 ailleurs[17]. Durant la période 2010-2013, on ne retrouve pas de signal de risque accru. Cela pourrait s'expliquer par le fait qu'en 2009-2010, la population vaccinée était d'une grandeur insuffisante pour détecter statistiquement une faible augmentation d'un événement rare.
Aujourd'hui, on estime que le risque de SGB après une grippe (17,2 cas pour 1 million de consultations pour grippe) est bien supérieur à celui de SGB après vaccination (1,03 pour 1 million de vaccinations)[17].
Aucun lien significatif n'a été retrouvé entre SGB et d'autres vaccinations[19].
Physiopathologie
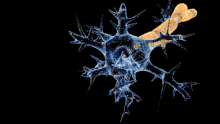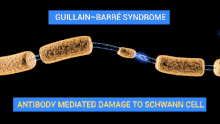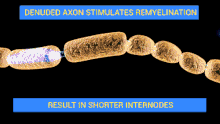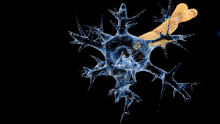
Les nerfs du patient sont attaqués par son propre système immunitaire (auto-immunité). Par exemple, certaines structures de la membrane externe de la bactérie Campylobacter jejuni peuvent ressembler à des structures présentes sur les nerfs, c'est le mimétisme moléculaire. En se défendant contre la bactérie, l'organisme attaque ses propres nerfs par des auto-anticorps (anticorps dirigés contre soi). Si l'on assimile un nerf à un câble électrique, l'attaque peut se porter sur la gaine (en l'occurrence la gaine de myéline), c'est la forme la plus fréquente des pays occidentaux[20]). Plus rarement l'axone (le "fil conducteur") est lui-même atteint, cette forme étant plus fréquente en Asie[9]. Selon les cas, la conduction nerveuse est ralentie, modifiée, voire interrompue.
Les SGB sont ainsi divisés en plusieurs sous-types, selon les structures moléculaires ciblées, et la localisation des lésions [21]..
Signes et symptômes
Le syndrome se manifeste de manière sporadique (apparemment par hasard, de façon isolée). Il est imprévisible, pouvant survenir en pleine santé.
Le début se caractérise le plus souvent par une faiblesse, voire une paralysie, des extrémités inférieures. Il existe, en second plan, des troubles sensitifs à type d'hypoesthésie (baisse du sens du toucher) en « gants » et en « chaussettes », engourdissements, fourmillements ou douleurs. Les réflexes ostéo-tendineux sont diminués ou abolis.
La maladie comporte trois phases d'évolution :
- Phase d'extension : elle correspond à une apparition rapide, de un jour à quatre semaines[6] d'une faiblesse, pouvant conduire à une paralysie franche, totale ou partielle. Le déficit est symétrique (affecte les deux côtés du corps à peu près de la même façon). Il est typiquement ascendant pour toucher les jambes, ensuite les bras, et parfois même les muscles respiratoires et le visage.
- Phase de plateau : elle dure de quelques jours à plusieurs mois, en fonction de la gravité des symptômes. Généralement, plus les déficits sont importants, plus cette phase de plateau est longue.
- Phase de récupération : elle dure plusieurs semaines, parfois des mois, elle se fait à l'inverse de la phase d'extension.
Une paralysie aiguë du nerf facial , parfois bilatérale, peut survenir dans la moitié des cas. Une atteinte transitoire du système nerveux central se voit dans un tiers des cas, caractérisée par des hallucinations, une psychose aiguë ou des troubles du sommeil, en règle régressive[22].
Plus rarement, des formes atypiques peuvent survenir, comme l'apparition de douleurs dans la ceinture lombaire se propageant assez vite dans les fesses puis les jambes, faisant croire à une sciatique touchant simultanément les deux membres inférieurs. L'usage des jambes devient vite incontrôlé et conduit à une paralysie plus ou moins totale des membres inférieurs. Ce tableau "descendant" peut se limiter à une atteinte de ces seuls membres inférieurs[23].
Dans la majorité des cas (80 %), les personnes atteintes récupèrent leurs capacités physiques (voire séquelles légères) au bout de 6 à 12 mois. Pour les 20 % restants, 5 % d'entre eux ont des séquelles lourdes et définitives (lorsque l'axone est touché) la motricité se retrouve altérée (on parle de maladie neuromusculaire). Un taux de mortalité est constaté entre 10 à 15 % (service de réanimation).
Les douleurs précédent les paralysies dans un tiers des cas et peuvent persister au-delà de l'amélioration des déficits, après plus d'un an d'évolution[24]. Une fatigue peut, de même, persister après guérison de l'atteinte motrice, quel que soit le niveau d'atteinte initial[25].
Diagnostic
Le diagnostic est principalement clinique, basé sur la répétition de l'examen neurologique montrant l'allure extensive ascendante, la faiblesse motrice symétrique, la diminution des réflexes.
Le diagnostic est confirmé par la ponction lombaire qui peut montrer, à partir de la 2e semaine, une augmentation de la concentration en protides dans le liquide céphalo-rachidien avec un compte normal de cellules du liquide céphalo-rachidien (dissociation albumino-cytologique). C'est l'élément de diagnostic le plus discriminant, mais ne permet pas un pronostic (son constat, précoce ou tardif, ne permet pas de préjuger la gravité de la maladie).
L'électromyographie consiste en la mesure de la vitesse de conduction nerveuse (VCN-EMG). Elle met en évidence un ralentissement ou une absence de conduction nerveuse dans près de 85 % des cas[20]. L'examen révèle s'il y a atteinte de l'axone des nerfs périphériques dans les cas les plus graves de SGB. La répétition de cet examen permet de préciser le sous-type de SGB en donnant des éléments de pronostic.
Le diagnostic différentiel doit éliminer d'autres causes de neuropathie périphérique (dans les cas où le SGB se présente d'abord de façon incomplète ou atypique). En premier lieu les causes mécaniques (compression médullaire), puis inhalation d'un solvant organique, absorption de plomb ou de certains médicaments, tels que la nitrofurantoïne ou la dapsone), les causes infectieuses (diphtérie, poliomyélite, botulisme), les causes métaboliques (porphyrie intermittente aiguë, hypokaliémie).
Complications
La gravité de la pathologie peut varier considérablement d'un cas moyen pouvant même ne pas être porté à la connaissance d'un médecin, à celui d'une maladie dévastatrice liée à une paralysie presque totale plaçant le patient entre la vie et la mort.
Il existe quatre principales complications du SGB qui justifient que le patient soit dans certains cas hospitalisés dans une unité de réanimation :
- détresse respiratoire : C'est la principale cause de mortalité. Celle-ci peut-être due à une paralysie du diaphragme, à une embolie pulmonaire, à une atéléctasie ou à une infection pulmonaire. Il est donc parfois nécessaire d'intuber le patient et de le placer sous ventilation assistée.
- troubles de la déglutition : due à une paralysie des muscles du pharynx. Il est alors requis l'installation d'une sonde naso-gastrique permettant une alimentation entérale ;
- dysautonomie : elle correspond à un dérèglement du système nerveux autonome (ou neurovégétatif) pouvant, dans les cas graves, conduire à un arrêt cardiaque. Il est donc important de surveiller les constantes cardiaques du patient (fréquence cardiaque et tension artérielle au minimum) et si nécessaire de traiter la bradycardie (atropine) ou l'hypotension (remplissage vasculaire) ;
- maladie thrombo-embolique veineuse : il faut à la fois prévenir les complications de décubitus et parfois instaurer un traitement anticoagulant.
Il existe également des complications ne mettant pas en jeu le pronostic vital du patient mais occasionnant une gêne importante, ce sont les rétractions tendineuses dues à l'absence de mouvements, elles peuvent être prévenues grâce à la kinésithérapie ; les douleurs doivent être prises en charge par des antalgiques spécifiques
Formes cliniques
Plusieurs formes se distinguent selon l'étude électromyographique. La forme habituelle est myélinique (démyélinisante) et guérit le plus souvent en quelques semaines sans séquelle. La forme axonale (c'est-dire par lésion de l'axone) est plus rare mais aussi plus sévère et peut laisser des séquelles à long terme, en touchant soit uniquement les nerfs moteurs, soit les nerfs moteurs et sensitifs. On parle alors de forme AMAN (Acute Motor Axonal Neuropathy)
Syndrome de Miller-Fisher
Le syndrome de Miller-Fisher, ou syndrome de Fisher, ou SMF, est une variante du syndrome de Guillain-Barré, caractérisée par :
- un manque de coordination des mouvements volontaires (ataxie) ;
- une absence de réflexes ;
- une paralysie des muscles moteurs des yeux (ophtalmoplégie).
Il a été décrit en 1955 pour la première fois[26]. L'évolution est, en règle générale, favorable et sans séquelle[27].
Autres formes
Formes brachio-cervico-pharyngées. Ce sont des formes localisées de SGB : sphère oro-pharyngées, muscles du cou et des épaules..
Pandysautonomies. Ce sont des atteintes exclusives du système nerveux autonome
Traitement
La prise en charge dans un centre de soins aigus est souhaitable, avec selon les cas, une convalescence dans un centre de rééducation, et un suivi par un programme de rééducation hors hôpital.
Les plasmaphérèses
Proposées dès 1978, les plasmaphérèses (échanges ou soustractions plasmatiques) ont été utilisées dans des cas graves du syndrome de Guillain-Barré avec une efficacité démontrée[28].
Les gammaglobulines
En 1988 et 1989, quelques chercheurs ont rapporté les effets bénéfiques de doses élevées de gammaglobulines (ou immunoglobulines) dans le traitement de la maladie. Ce traitement serait au moins aussi efficace que la plasmaphérèse, tout en étant plus simple d'administration (absence de nécessité d'un équipement spécialisé et de personnel qualifié)[29].
Autres traitements
La cortisone n'a pas démontré d'efficacité[9], même sur les douleurs[30].
La plupart des autres traitements ont pour but de prévenir ou de traiter les complications du syndrome : embolie pulmonaire (anticoagulants, bas de contention[31]). la lutte contre la constipation et l'infection urinaire secondaire à une rétention et à l'alitement prolongé doit être mise en œuvre.
Les douleurs peuvent nécessiter l'emploi de médicaments morphiniques.
L'indication de la mise sous respirateur artificiel dépend de la gravité de l'atteinte, la décision pouvant être aidée par l'objectivation d'une forte diminution de la capacité vitale mesurée lors d'épreuves fonctionnelles respiratoires[32].
Un traitement antibiotique ou antiviral ciblé sur le germe suspecté comme responsable est inutile, ce dernier ayant, en règle générale, disparu de l'organisme lorsque les premiers signes apparaissent.
Dans tous les cas, l'immobilisation entraîne un risque de phlébite des membres inférieurs qui doit être prévenu par l'administration d'héparine ou d'une contention élastique
Pronostic
L'évolution est difficile à prévoir. Les éléments défavorables sont : l'âge (plus de 50 ans), l'importance du déficit au début, l'atteinte respiratoire, l'importance des troubles à l'électromyogramme.
Le quart des patients requiert une ventilation assistée en raison de la faiblesse des muscles respiratoires[33].
La mortalité est d'un peu moins de 10 %[33]. Elle est essentiellement secondaire à des troubles du rythme cardiaque, une infection ou à une embolie pulmonaire[34]. Pour les formes sévères (troubles de la déglutition et paralysie des muscles respiratoires nécessitant une ventilation mécanique), elle atteint 20 %[35].
Il existe un risque de séquelles à long terme : de 5 à 15 % des patients demeureront invalides à plus ou moins long terme. 35 % environ se plaignent d'anomalies légères à long terme comme des étourdissements. La récidive est rare mais possible[36].
Malades célèbres
On pense aujourd'hui que c'est de séquelles de cette maladie que souffrait le président américain Franklin D. Roosevelt, paralysé des jambes à partir des années 1920, et non de la poliomyélite, comme avancé à l'époque[37],[38]. Autres célébrités :
- Markus Babbel, footballeur international allemand empêché de disputer la saison 2001-2002[39].
- Scott McKenzie, interprète en 1967 de la chanson San Francisco (Be Sure to Wear Flowers in Your Hair), décédé le des suites de cette maladie[40].
- Yasuoka Rikiya, acteur et chanteur japonais diagnostiqué en juin 2006, décède d'un arrêt cardiaque le 8 avril 2012[41].
- Mike Egener, joueur professionnel de hockey sur glace canadien[42].
- André Vingt-Trois, cardinal archevêque de Paris[43].
- Patrick Eaves, joueur professionnel de hockey sur glace canadien[44].
Témoignages
- Claude Pinault a été atteint d'un syndrome de Guillain-Barré de forme axonale sévère. De cette maladie, il en a tiré un récit Le Syndrome du bocal, qui a reçu le prix Paroles de patients 2009, publié chez Buschet Chastel[45].
- Philippe Valdenaire a été atteint d'un syndrome de Guillain-Barré qui s'est transformé ensuite en Polyradiculonévrite chronique. Il a fait paraître un ouvrage en septembre 2013 intitulé S’il vous plait... Merci avec le sous-titre Endormi par la maladie, celle-ci m’a réveillé publié chez L'atelier de la mémoire[46].
Voir aussi
Articles connexes
- Maladie autoinflammatoire
- Maladie auto-immune
- Vaccination
- Épidémiologie
- Syndrome de Bickerstaff
- Syndrome de Miller-Fisher
Liens externes
- [PDF] « Le syndrome de Guillain-Barré », ansm (consulté le 24 mai 2017)
- [PDF] « Le syndrome de Guillain-Barré », santé-sports.gouv.fr (consulté le 13 novembre 2009)
- « Le Syndrome de Guillain-Barré » (consulté le 16 novembre 2009). Ce site internet belge propose un groupe de soutien aux malades et à leurs proches.
- « Syndrome de Guillain-Barré : informations et témoignages ». Depuis 2000, ce site internet français propose de nombreux témoignages et échanges autour de la maladie.
- GBS/CIDP Foundation International
Notes et références
- Auguste-François Chomel, « De l'épidémie actuellement régnante à Paris », Journal hebdomadaire de Médecine, Paris « 1 », no 9, , p. 331-338
- Olivier Walusinski, « Octave Landry 1826-1865 », sur http://baillement.com, (consulté le 12 janvier 2014)
- Guillain G, Barré JA, Strohl A., « Sur un syndrome de radiculonévrite avec hyperalbuminose du liquide céphalo-rachidien sans réaction cellulaire: remarques sur les caractères cliniques et graphiques des réflexes tendineux », Bulletins et mémoires de la Société des Médecins des Hôpitaux de Paris, no 40, , p. 1462-70
- (en) J.B Winer, « An update in Guillain-Barré Syndrome », Autoimmune Diseases,
- Guillain G., « Synthèse générale... », Journal belge de Neurologie et Psychiatrie, no 4,
- (en) Yuki N, Hartung HP, « Guillain–Barré syndrome » N Engl J Med. 2012;366:2294-2304
- (en) A.K. Asbury, « Criteria for diagnosis of Guillain-Barre Syndrome », Ann. Neurol., no 3, , p.565-566
- (en) Sejvar JJ, Baughman AL, Wise M, Morgan OW, « Population incidence of Guillain-Barré syndrome: a systematic review and meta-analysis » Neuroepidemiology 2011;36:123-133
- (en) Winer JB, « Guillain-Barré syndrome » BMJ 2008;337:a671.
- (en) A.D Walling, « Guillain-Barré Syndrome », American Family Physician, no 3,
- Campylobacter Species and Guillain-Barré Syndrome lire en ligne
- E. Garcia, « Zika virus infection », Relevé épidémiologique hebdomadaire, no 7, , p.77
- Affsaps : Le syndrome de Guillain-Barré
- M. Gregg, « Le pouvoir de l'épidémiologie dans la pratique de la santé publique », Bulletin Epidémiologique Hebdomadaire, no 14,
- (en) D.J Sencer, « Reflections on the 1976 Swine Flu Vaccination Program », Emerging Infectious Diseases, no 1, , p.29-33
- Influenza Vaccines and Neurological complications. Immunization Review. Institute of Medicine. 6 octobre 2003
- (en) C. Vellozzi, « Guillain-Barré Syndrome, Influenza, and Influenza vaccination », Clinical Infectious Diseases, no 58,
- Salmon DA, Proschan M, Forshee R et al. Association between Guillain-Barré syndrome and influenza A (H1N1) 2009 monovalent inactivated vaccines in the États-Unis: a meta-analysis, Lancet, 2013;381:1461-1468
- (en) R. Baxter, « Recurrent Guillain-Barré Syndrome Following Vaccination », Clinical Infectious Diseases, no 54,
- (en) Hadden RD, Cornblath DR, Hughes RA et al. « Electrophysiological classification of Guillain-Barré syndrome: clinical associations and outcome. Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group » Ann Neurol. 1998;44:780-8
- (en) Yuki N, « Molecular mimicry between gangliosides and lipopolysaccharides of Campylobacter jejuni isolated from patients with Guillain-Barré syndrome and Miller Fisher syndrome » J Infect Dis. 1997;176(suppl 2):S150-3.
- (en) Cochen V, Arnulf I, Demeret S et al. « Vivid dreams, hallucinations, psychosis and REM sleep in Guillain-Barré syndrome » Brain 2005;128:2535-2545.
- « forme descendante (phase d'extension page 3) »
- (en) Ruts L, Drenthen J, Jongen JL et al. « Pain in Guillain-Barré syndrome: a long-term follow-up study » Neurology 2010;75:1439-1447.
- (en) Garssen MP, van Koningsveld R, van Doorn PA, « Residual fatigue is independent of antecedent events and disease severity in Guillain-Barré syndrome » J Neurol. 2006;253:1143-1146.
- (en) M Fisher, « An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and areflexia) », N Engl J Med., no 255, , p. 57-65 (lire en ligne)
- (en) Mori M, Kuwabara S, Fukutake T, Yuki N, Hattori T., « Clinical features and prognosis of Miller Fisher syndrome », Neurology, no 56, , p. 1104-1106. (lire en ligne)
- (en) Raphael JC, Chevret S, Hughes RA, Annane D., « Plasma exchange for Guillain-Barré syndrome », Cochrane Database Syst Rev, no 2, , CD001798 (lire en ligne)
- Hughes RA, Raphael JC, Swan AV, van Doorn PA, Intravenous immunoglobulin for Guillain-Barré syndrome, Cochrane Database Syst Rev, 2001;(2):CD002063
- (en) Ruts L, van Koningsveld R, Jacobs BC, van Doorn PA, « Determination of pain and response to methylprednisolone in Guillain-Barré syndrome », J Neurol., no 254, , p. 1318-1322 (lire en ligne)
- (en) Hughes RAC, Wijdicks EF, Benson et al., « Supportive care for patients with Guillain-Barré syndrome », Arch Neurol., no 62, , p. 1194-1198 (lire en ligne)
- (en) Lawn N, Fletcher D, Henderson R, Wolter T, Wijdicks E., « Anticipating mechanical ventilation in Guillain-Barré syndrome », Arch Neurol., no 58, , p. 893-8. (résumé)
- (en) Rees JH, Thompson RD, Smeeton NC, Hughes RA., « Epidemiological study of Guillain-Barré syndrome in south east England », J Neurol Neurosurg Psychiatry, no 64, , p. 74-7
- (en) Winer JB, Hughes RA, Osmond C., « A prospective study of acute idiopathic neuropathy. I. Clinical features and their prognostic value », J Neurol Neurosurg Psychiatry, no 51, , p. 605-12 (résumé)
- T. Sharshar, S. Siami et D. Orlikowski, « Ce malade est-il atteint d’un syndrome de Guillain-Barré ? », Réanimation, vol. 16, no 6, , p. 504-510
- (en) Kuitwaard K, van Koningsveld R, Ruts L, Jacobs BC, van Doorn PA., « Recurrent Guillain-Barré syndrome », J Neurol Neurosurg Psychiatry, no 80, , p. 56-59 (lire en ligne)
- (en) Goldman AS, Schmalstieg EJ, Freeman DeH, Goldman DA, Schmalstieg FC, « What was the cause of Franklin Delano Roosevelt's paralytic illness? », J Med Biogr, vol. 11, no 4, , p. 232–40 (PMID 14562158, lire en ligne [PDF])
- http://news.sciencemag.org/2003/10/did-fdr-have-guillain-barr%C3%A9
- http://www.down-the-line.com/the-era-of-the-great-marcus-babbel.html
- Le chanteur de "San Francisco", Scott McKenzie, est mort sur Le Nouvel Observateur, 20 août 2012
- L'acteur Yasuoka Rikiya est mort d'après l'université de Californie du Sud, 9 avril 2012
- (en) « Coventry Blaze: Canadian captain Mike Egener will not be back this season due to serious illness », sur Coventry Telegraph, (consulté le 28 juillet 2015).
- « Absence du cardinal Vingt-Trois »
- Modèle {{Lien web}} : paramètre «
titre» manquant. (en) http://www.sportsnet.ca/hockey/nhl/ducks-patrick-eaves-recovering-guillain-barre-syndrome/, sur Sportnet (consulté le 5 janvier 2018) - Claude Pinault en parle sur son site web www.claudepinault.fr
- Philippe Valdenaire en parle sur son site web www.philippevaldenaire.fr

