Biologie structurale
La biologie structurale est la branche de la biologie qui étudie la structure et l'organisation spatiale des macromolécules biologiques, principalement les protéines et les acides nucléiques. La biologie structurale concerne en particulier la détermination à l'échelle atomique de la structure 3D au moyen de techniques biophysiques, les principes à la base des modifications de conformation des macromolécules, l'analyse des mouvements moléculaires et la dynamique de ces structures.
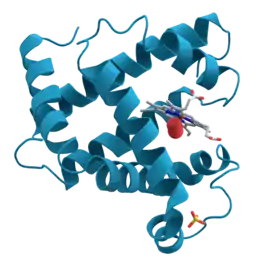
Les données issues de la biologie structurale sont fréquemment utilisées dans les projets de développement de médicaments. En effet, la compréhension moléculaire et structurale du mode d'action d'un médicament sur sa cible, visualisée par ces approches, permet de rationaliser la conception et l'amélioration de molécules actives. On utilise pour cela à la fois des méthodes expérimentales d'analyse structurale et des approches computationnelles pour prédire des candidats médicaments à partir de la structure.
Techniques de biologie structurale
Les techniques de microscopie optique ne disposent pas de la résolution suffisante pour distinguer les détails des molécules biologiques au niveau atomique. Les outils de la biologie structurale sont le plus souvent des méthodes physiques indirectes. La principale méthode est la cristallographie aux rayons X, historiquement la plus ancienne et toujours la plus utilisée. Elle est complétée par la résonance magnétique nucléaire, qui permet aussi d'atteindre la résolution atomique ainsi que d'obtenir des informations sur la dynamique des molécules en solution. D'autres techniques sont aussi utilisées, comme le diffusion des rayons X aux petits angles et la diffusion des neutrons (pratiquée en France à l'Institut Laue-Langevin), ces méthodes ne permettent pas d'obtenir des informations à haute résolution mais permettent d'étudier des molécules en solution sans les limitations des autres méthodes (nécessité d'obtenir des monocristaux ayant un pouvoir diffractant pour la cristallographie ; limitations a des échantillons de petite taille pour la RMN).
La cryo-microscopie électronique, du fait de nouveaux détecteurs[2] et de développements dans le traitement de données, a significativement amélioré les résolutions atteintes en routine[3]. Ne souffrant pas des limitations de la cristallographie et de la RMN, la cryo-microscopie électronique est en train de remplacer la cristallographie comme principale technique de la biologie structurale pour les complexes macromoléculaires de grande taille.
Cristallographie des macromolécules

La résolution de la structure cristallographique est basée sur l'étude de la diffraction des rayons X par des cristaux de macromolécules. Les rayons X sont diffractés par les électrons et l'analyse permet de remonter à la densité électronique de la molécule, c'est-à-dire la localisation des électrons qui gravitent autour d'elle. Cette densité, pourvu qu'elle soit suffisamment précise, permet de remonter à la localisation de chaque atome de la molécule et donc à ses coordonnées cartésiennes.

Le pré-requis obligatoire pour ce type d'étude est l'obtention de cristaux. Dans des conditions physico-chimiques appropriées (pH, tampon, ions...), les macromolécules en solution peuvent en effet former des cristaux contenant plus ou moins de solvant aqueux. La recherche de ces conditions physico-chimiques favorables est largement empirique, et variable d'une molécule à l'autre. Le degré de pureté de la molécule est également un facteur déterminant. Cette étape de cristallisation est donc souvent limitante dans ce type d'analyse.
Une fois des cristaux de taille suffisante obtenus (quelques dizaines à quelques centaines de microns), on les place dans un faisceau de rayons X monochromatiques, produit par une source adaptée, un générateur de laboratoire ou un synchrotron, ces derniers produisant des faisceaux nettement plus intenses. Dans ces conditions, les électrons des molécules ordonnées dans le cristal diffractent les rayons X. Avec un détecteur, on enregistre des images du motif de diffraction obtenu qui est constitué de taches régulièrement espacées, pour différentes orientations du cristal. L'espacement des tâches renseigne sur la maille du cristal, c'est-à-dire les dimensions de l'élément de volume qui est régulièrement répété dans l'espace, pour donner naissance au cristal. Les intensités des tâches contiennent quant à elles une partie de l'information sur la densité électronique de chacune des macromolécules présentes dans la maille. La phase du signal associé à chaque tache est cependant perdue et il faut alors la reconstituer. Différentes méthodes permettent de reconstruire les phases :
- Le remplacement isomorphe multiple, qui consiste à faire diffuser des atomes lourds (riches en électrons) dans le cristal et à comparer la diffraction avec et sans ces éléments lourds. La présence de l'élément lourd modifie légèrement les intensités de diffraction, ce qui permet de calculer les phases par triangulation, pourvu que l'on puisse positionner les atomes lourds dans la maille du cristal ;
- La diffusion anomale, qui consiste à faire varier la longueur des rayons X autour du seuil d'absorption d'un des types d'atomes contenu dans la molécule. Le sélénium est fréquemment utilisé car il possède un seuil d'absorption X proche des longueurs d'onde utilisées (autour de 0,1 nm). Pour les protéines, on introduit en général des acides aminés séléniés : sélénocystéine ou sélénométhionine par voie biosynthétique ;
- Le remplacement moléculaire, qui est une méthode utilisant la structure d'une protéine homologue lorsque celle-ci est connue. On essaye de placer la structure homologue dans la maille du cristal et on calcule la diffraction théorique que l'on compare à celle qui est observée. Ceci permet de calculer des phases théoriques initiales ;
Une fois les phases reconstruites, on peut remonter à la densité électronique par transformation de Fourier des signaux de diffraction. On peut alors reconstruire la molécule dans la densité électronique en utilisant des systèmes automatiques ou des logiciels graphiques interactifs, ce qui permet d'obtenir les coordonnées cartésiennes de chaque atome de la molécule. À partir du modèle construit, on peut calculer des intensités de diffraction que l'on compare aux intensités mesurées expérimentalement, ce qui permet d'améliorer progressivement le modèle, une étape appelée "affinement".
RMN des macromolécules

La résonance magnétique nucléaire ou RMN utilise les propriétés magnétiques des noyaux des atomes pour obtenir des informations géométriques (distances, angles...) sur la molécule. À partir de ces informations géométriques, il est ensuite possible d'en reconstruire par le calcul une structure 3D, par une sorte de triangulation.
Les noyaux de certains atomes possèdent un spin nucléaire, en particulier l'hydrogène 1H, le carbone 13C et l'azote 15N. À ce spin est associé un moment dipolaire magnétique. Placés dans un champ magnétique très intense (typiquement 10 à 20 tesla), ces spins s'alignent avec le champ. La RMN étudie les interactions entre spins dans une molécule et permet d'observer principalement deux types d'effets :
- Les interactions scalaires, qui sont des couplages médiés par les électrons de liaison. Ces interactions scalaires permettent d'identifier les connexions à travers les liaisons covalentes de la molécule et donc d'en reconstruire ou d'en identifier la topologie. Elles dépendent de la géométrie des liaisons et en particulier des angles dièdres entre les atomes ;
- Les interactions dipolaires qui correspondent à un transfert d'aimantation à travers l'espace, appelé effet Overhauser nucléaire. Ces interactions dipôle magnétique-dipôle magnétique ne dépendent que de la distance entre les atomes et pas de la géométrie des liaisons et sont observables jusqu'à environ 0,5 nm ;
Un projet de RMN biologique structurale va utiliser un échantillon en solution de la macromolécule et enregistrer des spectres contenant des corrélations des deux types ci-dessus (scalaires et dipolaires). À partir de ces spectres, on va réaliser l'analyse des différents signaux, qui vont être individuellement attribués à chaque atome de la molécule. Cette phase s'appelle l'attribution spectrale. Ensuite, on va collecter un ensemble de données géométriques sur la molécule (distances inter-atomiques, angles dièdres...), appelées contraintes. À partir de ces informations géométriques locales, on va reconstruire par le calcul une structure 3D compatible avec ces contraintes : la structure RMN en solution[4]. Pour cela, on part d'une conformation aléatoire de la molécule, à laquelle on ajoute progressivement les contraintes pour induire son repliement, à l'aide des contraintes. Ce processus est répété à partir de plusieurs conformations initiales différentes, afin de vérifier que le calcul converge vers une solution unique. Pour cette raison, les structures RMN de macromolécules sont en général constituées d'un ensemble de conformations proches.
Cryomicroscopie électronique

La cryomicroscopie électronique permet de reconstituer la structure à basse résolution de complexes macromoléculaires. Un échantillon en solution de l'objet à étudier est soumis à une congélation "flash", par immersion rapide dans un liquide cryogénique (éthane liquide maintenu dans un bain d'azote liquide). Dans ces conditions, l'eau constituant le solvant est vitrifiée en une structure amorphe, qui préserve la forme des macromolécules qu'elle contient.
On réalise alors un cliché de microscopie électronique à transmission, dans lequel vont figurer un grand nombre d'images de la molécule d'intérêt, vue sous les différentes orientations dans laquelle elle a été prise dans la glace. Au moyen d'outils informatiques, on va repérer et classifier ces différentes images, afin de regrouper celles qui se ressemblent et correspondent à une vue de l'objet biologique étudié sous une orientation similaire. Sur chacune de ces classes, on va ensuite effectuer des opérations de sommation/moyenne des images ainsi que de filtrage du bruit de fond, pour améliorer le rapport signal sur bruit.
À l'issue de ce traitement, on obtient une série de vues 2D précises de la molécule, qui correspondent à des projections de sa structure 3D. À nouveau au moyen d'outils informatiques, on va alors reconstruire une enveloppe moléculaire en trois dimensions à partir de ces différentes images 2D. Plusieurs milliers d'images individuelles sont en général nécessaires pour obtenir une reconstruction précise, qui permettra d'avoir une surface d'enveloppe de l'objet, plus ou moins détaillée. Typiquement, pour des édifices non-symétriques, la précision de ces reconstructions est de l'ordre de 1 à 2 nanomètres, parfois subnanométrique pour les systèmes les mieux définis.

Dans le cas d'objets qui sont des oligomères symétriques, comme des capsides de virus, on va de plus utiliser l'information de symétrie intrinsèque de l'objet pour améliorer la précision de sa reconstruction, en imposant cette symétrie lors de la reconstruction de l'enveloppe moléculaire.
Méthodes de diffusion
Les méthodes de diffusion au petits angles sont des techniques permettant d'obtenir des informations sur la taille et la forme d'objets contenus dans une solution. En biologie, elle permet d'obtenir des informations à basse résolution sur l'état d'oligomérisation et la forme des macromolécules biologiques[5]. Deux principales techniques de diffusions aux petits angles sont utilisées en biologie structurale : la diffusion des rayons X (SAXS : small angle X-ray scattering) et la diffusion des neutrons (SANS : small angle neutron scattering').
Le principe de ces expériences est simple : une solution concentrée de la macromolécule à étudier est placée dans un faisceau parallèle de rayons X ou de neutrons. Les objets de la solution diffusent le rayonnement incident. Dans le cas des rayons X, la diffusion résulte de l'interaction entre le nuage électronique et les photons. Dans le cas des neutrons, la diffusion résulte de l'interaction avec le noyau des atomes de l'objet en solution. On collecte l'intensité du signal diffusé en fonction de l'angle par rapport au faisceau incident. Ce signal, qui résulte d'une moyenne sur toutes les orientations de la molécule étudiée, donne une information sur sa taille et sa forme ou "enveloppe". En fonction de l'échantillon, les techniques de diffusion permettent des reconstructions avec des précisions pouvant atteindre 1 nm de résolution, en particulier pour des objets possédant des symétries internes (enveloppes de virus, protéines multimériques...).
Modélisation moléculaire
La modélisation moléculaire est un ensemble de méthodes à la frontière de la biologie structurale et de la bio-informatique. Elle consiste à effectuer des calculs informatiques pour reconstruire les structures, effectuer des prédictions structurales[6] ou analyser des propriétés dynamiques de macromolécules. Elle repose souvent sur l'utilisation d'une description des forces moléculaires qui agissent sur les atomes pour simuler des mouvements ou trouver des conformations d'énergie minimale.
Base de données
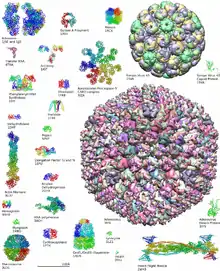
L'essentiel des données sur les structures des macromolécules biologiques sont aujourd'hui déposées sur une base de données publique, la Protein Data Bank ou PDB[7]. La PDB contient, sous un format standardisé, les données structurales sur les coordonnées des atomes de plus de 100 000 macromolécules ou complexes (chiffres de ). Ces données sont accessibles librement en ligne (voir liens externes) et les structures peuvent être visualisées au moyen de divers outils logiciels interactifs, dont un grand nombre sont gratuits pour un usage académique.
Les structures PDB sont identifiées par un code unique à 4 lettres ou chiffres, dont le premier est en général un chiffre (exemple : l'identifiant PDB de la structure de la myosine du grand cachalot illustrant l'introduction de cet article est 1MBO[8]). La PDB recueille la majorité des structures publiées dans la littérature scientifique, le dépôt à la PDB constituant un pré-requis pour l'acceptation d'une publication par la plupart des journaux scientifiques.
Aujourd'hui, la majorité des structures de macromolécules biologiques déterminées par cristallographie aux rayons X déposées dans la PDB provient de données obtenues à l'aide de rayons X générés à partir de rayonnement synchrotron. L'utilisation de synchrotrons dans la détermination tridimensionnelle de macromolécules biologique a contribué à environ 80 % des enregistrements depuis 1995 dans la PDB[9].
Applications de la biologie structurale
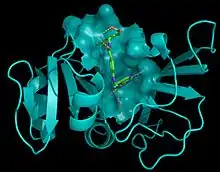
La biologie structurale a des applications importantes dans le domaine de la biochimie et de l'enzymologie, pour la compréhension des mécanismes catalytiques et de la reconnaissance spécifique des substrats. L'une des plus importantes applications de la biologie structurale concerne la conception rationnelle de médicaments ou la structure tridimensionnelle de la cible (enzyme, récepteur) et est utilisée pour aider à concevoir les ligands. Dans certains cas, on peut résoudre la structure de la cible en complexe avec un ou des ligands fixés, ce qui permet de comprendre les bases moléculaires de la reconnaissance et utiliser cette information pour en améliorer ou en modifier les propriétés (affinité, activité, sélectivité...).
Notes et références
- (en) J.C. Kendrew, G. Bodo, H.M. Dintzis, R.G. Parrish, H. Wyckoff et D.C. Phillipps, « A three-dimensional model of the myoglobin molecule obtained by x-ray analysis », Nature, vol. 181, , p. 662-666 (PMID 13517261)
- (en) « Direct Electron Detectors », Methods in Enzymology, vol. 579, , p. 1–17 (ISSN 0076-6879, DOI 10.1016/bs.mie.2016.05.056, lire en ligne, consulté le )
- Eva Nogales, « The development of cryo-EM into a mainstream structural biology technique », Nature methods, vol. 13, no 1, , p. 24–27 (ISSN 1548-7091, PMID 27110629, PMCID PMC4913480, lire en ligne, consulté le )
- Frédéric Dardel et Thérèse Maillavin, « Structure des protéines par RMN », Techniques de l'ingénieur, vol. TI053, , AF6608 (lire en ligne)
- (en) D.I. Svergun et M.H.J. Koch, « Small-angle scattering studies of biological macromolecules in solution », Rep. Prog. Phys., vol. 66, , p. 1735–82 (DOI 10.1088/0034-4885/66/10/R05, Bibcode 2003RPPh...66.1735S)
- Thomas Simonson, « Le « problème du repliement » Peut-on prédire la structure des protéines ? », Médecine/Sciences, vol. 21, , p. 609-612 (PMID 15985203, lire en ligne)
- (en) H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov et P.E. Bourne, « The Protein Data Bank », Nucleic Acids Res., (PMID 10592235, DOI 10.1093/nar/28.1.235)
- S. E. Phillips, « Structure and refinement of oxymyoglobin at 1.6 A resolution », Journal of Molecular Biology, vol. 142, no 4, , p. 531–554 (ISSN 0022-2836, PMID 7463482, lire en ligne, consulté le )
- « Statistics by Year : BioSync », sur biosync.sbkb.org (consulté le )
- (en) D.A. Matthews, R.A. Alden, J.T. Bolin, S.T. Freer, R. Hamlin, N. Xuong, J. Kraut, M. Poe, M. Williams et K. Hoogsteen, « Dihydrofolate reductase: x-ray structure of the binary complex with methotrexate. », Science, vol. 197, , p. 452-455 (PMID 17920, DOI doi:10.1126/science.17920)
Voir aussi
Bibliographie
Articles connexes
Liens externes
- Protein Data Bank en Europe, base de données internationale de biologie structurale
- Protein Data Bank en Japan, base de données internationale de biologie structurale
- Le Collectif de recherche pour Bioinformatique structurale aux États-Unis
- Proteopedia, wiki permettant une visualisation interactive des structures de la PDB
- PDB-101: Molecule of the Month: Myoglobin
 Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire