Paralysie supranucléaire progressive
La paralysie supranucléaire progressive (PSP ou maladie de Steele-Richardson-Olszewski, du nom des médecins qui l'ont caractérisée en 1963), est une maladie neurogénérative qui est due à la destruction progressive des neurones de différentes régions du cerveau : le striatum, la formation réticulée du tronc cérébral, le locus niger, les noyaux des nerfs crâniens. Ces lésions entraînent notamment une difficulté à effectuer des saccades oculaires verticales (ophtalmoplégie supranucléaire), caractéristique de la PSP, mais elles affectent aussi progressivement l’équilibre, la vue, la marche, la déglutition, la parole et d'une manière plus générale, les mouvements effectués dans le plan sagittal. La PSP touche environ six personnes sur 100 000, autant les hommes que les femmes, sans qu'on ait pu identifier de facteur de risque lié à la race, l'environnement de vie ou l'activité professionnelle. Certains des symptômes de la PSP se rapprochent de ceux de la maladie de Parkinson ou de l'atrophie multisystématisée mais elle s'en distingue par les atteintes oculomotrices. Les mécanismes pathologiques de la PSP restent mal connus : on la décrit parfois comme une taupathie car la dégénrescence neurofibrillaire est associée à une accumulation de protéine Tau dans les cellules nerveuses.

| Spécialité | Neurologie |
|---|
| CIM-10 | G23.1 |
|---|---|
| CIM-9 | 333.0 |
| OMIM | 601104 |
| DiseasesDB | 10723 |
| MedlinePlus | 000767 |
| eMedicine | 1151430 |
| eMedicine | neuro/328 |
| MeSH | D013494 |
![]()
Découverte
Elle a été décrite pour la première fois, estime-t-on, en 1904, bien que des recherches d’antériorité évoquent des cas de PSP probable au moins au milieu du XIXe siècle[1]. Ce n’est pourtant que suite aux observations du Pr J. Clifford Richardson durant les années 1950 à Toronto, et après qu’il eut invité le Pr John Steele et le Pr Jerzy Olszewski (pl) à étudier les changements pathologiques dans le système nerveux central, que la PSP sera identifiée comme une maladie à part entière, à partir de juin 1963[2]. Avec la publication en 1964 des travaux des trois professeurs neurologues, la PSP sera reconnue sous son nom actuel, comme une entité nosologique spécifique, connue également sous le nom de « maladie de Richardson-Olszewksi », « syndrome de Steele » aux États-Unis, et sous le nom de « maladie de Steele-Richardson-Olszewski » en Europe.
Le syndrome de Guam, également mis au jour par le Pr John Steele, est une forme particulière de PSP de l'île de Guam, dans le Pacifique. C'est donc aussi un syndrome parkinsonien atypique qui est souvent l'association d'une maladie de Parkinson à une Sclérose Latérale Amyotrophique et à une démence.
Explication du terme
En général, une « paralysie » est une faiblesse motrice ou une paralysie d'une partie du corps.
Le terme « supranucléaire » se réfère à la nature du problème oculaire dans la PSP. Bien que certains patients atteints de PSP décrivent leur symptôme comme « flou », le problème réel est l'incapacité d’une visée correcte des yeux à cause d’une faiblesse (ou paralysie) des muscles qui font bouger les globes oculaires. Ces muscles sont contrôlés par des cellules nerveuses qui résident en grappes, ou « noyaux », près de la base du cerveau, dans le tronc cérébral.
La plupart des problèmes qui affectent les mouvements oculaires ont pour origine ces noyaux du cerveau, mais dans la PSP le problème provient de parties du cerveau qui contrôlent ces noyaux eux-mêmes. Ces zones de contrôle « plus élevées » sont ce que le préfixe « supra » désigne dans « supranucléaire » (au-dessus des noyaux).
Physiopathologie
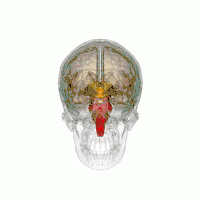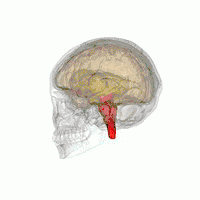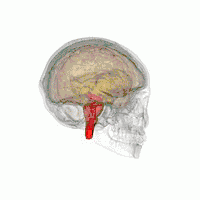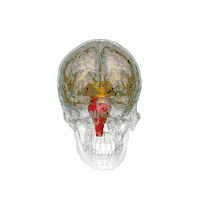
Les principales régions du cerveau affectées par la PSP sont les suivantes :
- le tronc cérébral, en particulier la partie du mésencéphale où réside le mouvement « supranucléaire » de l'œil ;
- les ganglions de la base, en particulier le noyau sous-thalamique, la substance noire (ou locus niger) et le globus pallidus ;
- le cortex cérébral, en particulier la zone des lobes frontaux ;
- le noyau dentelé du cervelet (le cervelet représente un bon 50 % de nos neurones) ;
- et la moelle épinière, en particulier la zone où sont commandés certains contrôles de la vessie et de l'intestin.
Épidémiologie
La PSP est une maladie rare car elle touche moins d'une personne sur 2 000, seuil en dessous duquel, dans l'Union Européenne, une maladie est définie comme rare. La prévalence la plus rapportée pour la PSP, est d'environ 6 malades pour 100 000 personnes. Ce chiffre ne demeure qu'une indication à ce jour. C'est également une maladie orpheline dans la mesure où il ne lui est pas connu actuellement de traitement efficace. La PSP est une maladie de pronostic sévère.
Elle affecte plusieurs dizaines de milliers de personnes aux États-Unis et en Europe (40 à 60 000, selon les estimations). Elle affecte un peu plus les hommes que les femmes (écart d'environ 2 %). Elle se déclare majoritairement chez des personnes âgées entre 55 et 65 ans.
Diagnostic

Symptômes
Les symptômes les plus précoces sont un ralentissement intellectuel avec une conservation de la mémoire épisodique, des modifications du comportement du type de la perte d'intérêt (apathie) ou de l'irritabilité. Ces troubles comportementaux peuvent mimer un état dépressif. Les patients consultent le plus souvent pour des troubles d'équilibre sévères, responsables de chutes. Ces patients peuvent tomber plusieurs fois par jour ; chutes en arrière, au demi-tour. Les patients sont très instables et ont tendance à se laisser tomber dans le fauteuil. La perte d'équilibre s'aggrave au point de rendre la marche difficile voire impossible.
Les troubles de la fluence verbale et de la parole sont variés : bégaiement (palilalie, écholalie), persévération verbale, nasillement et difficulté à émettre des phrases compréhensibles.
Trouble de l'attention : le patient a du mal à soutenir une conversation sur le long terme, changeant abruptement de sujet sans s'en apercevoir au bout de quelques minutes. Ce délai se raccourcit jusqu'à rendre la conversation impossible, avec une évolution concomitante vers une aphasie.
Aux troubles de l'équilibre et de la mobilité viennent s'ajouter des troubles plus insidieux tels que des difficultés visuelles. Ces problèmes visuels résultent d'une incapacité à guider les yeux en raison de l'atteinte des centres cérébraux contrôlant les mouvements oculaires. Les troubles de la déglutition sont plus tardifs, pouvant entraîner des « fausses routes » alimentaires, qui elles-mêmes entrainent des complications pulmonaires : l'asphyxie par étouffement est une fréquente cause de décès en fin d'évolution. Pour conserver une alimentation correcte au malade, sans fausse route, une gastrostomie peut devenir incontournable, tout comme l'emploi d'un aspirateur trachéal pour les mucosités.
L’ordre d’apparition des symptômes étant variable d’une personne à l’autre et les symptômes eux-mêmes étant communs à d’autres pathologies, le diagnostic demeure difficile. Le diagnostic peut être suspecté cliniquement devant des troubles posturaux, des signes parkinsoniens axiaux et des troubles oculomoteurs. L'examen clinique recherchera une lenteur dans le regard vers le haut, une rigidité cervicale, d'une manière plus générale un déficit moteur dans le plan sagittal, des troubles cognitifs sous-corticaux frontaux et une difficulté à arrêter un mouvement moteur répété comme une séquence de 3 applaudissements.
Le déficit moteur dans le plan sagittal explique les difficultés oculaires verticales, les chutes spontanées en arrière à l'arrêt par défaut de correction de la posture, les difficultés à déglutir. La déficience oculomotrice induit également des chutes en avant, le malade ne pouvant plus vérifier subrepticement la présence d'obstacles au sol sur son trajet (adaptation de l'environnement du malade).
Au nombre des principaux symptômes et signes cliniques, il faut retenir :
- les perturbations de l'équilibre allant jusqu'aux chutes (signe précoce) et plus particulièrement vers l’arrière,
- l'aimantation du regard : perte de la mobilité du regard et particulièrement à bouger les yeux du haut vers le bas,
- les modifications du comportement : ralentissement de la marche, de la fluidité verbale, apathie, impulsivité, agressivité, instabilité de l’attention et élocution monotone,
- le comportement de préhension : difficulté à desserrer la main des objets tenus,
- la présence d'un syndrome dysexécutif : persévérations, perturbation du jugement, difficultés à résoudre des problèmes.
Examens complémentaires
L'IRM cérébrale peut révéler quelques anomalies caractéristiques : amincissement de la partie supérieure de la protubérance (coupe sagittale), écartement des pédoncules cérébraux (coupe axiale), élargissement du 4e ventricule (coupe axiale), atrophie frontale.
D'une façon générale, c'est l'examen neuropathologique qui pose le diagnostic de PSP, en fonction de la distribution topographique des lésions, dont l'interprétation peut être commune aux autres taupathies telles que la DCB et certaines DFT de types lobaires.
À ce jour il a été établi cinq variantes de la PSP :
- la PSP classique (la plus fréquente) et quatre autres atypiques :
- la PSP de type parkinsonien (PSP-P),
- la PSP-akinésie pure avec « freezing » de la marche (PSP-PAGF),
- la PSP-syndrome corticobasal (PSP-CBS),
- la PSP-aphasie progressive non fluente (PSP-PNFA).
Diagnostic différentiel
Les diagnostics différentiels principaux sont :
- Maladie d'Alzheimer (MA)
- Maladie de Parkinson (MP)
- Dégénérescence Cortico-Basale (DCB)
- Dégénérescence Fronto-Temporale (DFT) dont la Maladie de Pick
- Atrophie Multi-Systématisée (AMS)
La PSP peut être difficile à reconnaître au moins en début de maladie car les troubles initiaux sont également présents dans d’autres maladies neurodégénératives, comme pour les plus connues celles de Parkinson et d'Alzheimer, mais aussi comme celles de l'AMS (Atrophie Multi-Systématisée), ou la DCB (Dégénérescence Cortico-Basale) qui est très proche de la PSP. Il n'y a pas si longtemps, dans le cas de la DCB, des médecins expérimentés se trompaient dans plus de 50 % des cas[3].
L'errance de diagnostic demeure encore importante, en moyenne plus de trois ans, avec de fortes disparités. On considère que la PSP représente au moins 5 % des syndromes parkinsoniens, ce qui lui vaut d'être en importance, le deuxième syndrome parkinsonien après la maladie de Parkinson.
L'ophtalmoplégie supranucléaire (« aimantation du regard », fixité et réduction du champ visuel surtout vertical) dont les malades sont atteints, demeure l'une des premières caractéristiques visibles pour le diagnostic. En effet, la conséquence de la paralysie des globes oculaires est d'entraîner des chutes inexpliquées et en arrière. Par contre il n'y a pas de tremblement parkinsonien et les symptômes sont résistants au traitement par la L-Dopa.
La PSP est un des syndromes parkinsoniens dont le processus évolutif est connu mais pas la cause exacte. Elle se distingue de la maladie de Parkinson par une évolution plus sévère, avec peu ou pas de réponse au traitement. La PSP, comme la DCB qui lui est très similaire, sont des maladies de mauvais pronostic, avec une évolution par paliers, très invalidantes et dont seuls les symptômes peuvent à ce jour être traités. Des soins palliatifs sont à prévoir.
Dans les syndromes parkinsoniens dégénératifs atypiques parfois qualifiés de syndromes parkinsoniens plus, qui se différencient de la maladie de Parkinson notamment par la faible, voire l’absence de réactivité au traitement dopaminergique résultant, entre autres, de lésions postsynaptiques, on distingue donc :
- l'atrophie multisystématisée et la démence à corps de Lewy dans le cadre des synucléinopathies ;
- la paralysie supranucléaire progressive et la dégénérescence corticobasale dans le cadre des tauopathies.
Génétique et facteurs de risque
La PSP n'est pas héréditaire. En effet, lorsqu'un membre d'une famille est atteint d'une PSP, il y a moins de 1 % de cas où d'autres membres de cette même famille présentent les symptômes d'une PSP. En comparaison, pour une maladie également considérée non-héréditaire : 15 % des personnes ayant un Parkinson ont un proche atteint[4], et au moins 5 % des malades sont reconnus comme ayant une forme de Parkinson liée à une mutation de gènes spécifiques[5].
Il a été liée à la PSP une variante du gène de la protéine tau appelé l'haplotype H1, situé sur le chromosome 17. Presque toutes les personnes atteintes de PSP ont reçu cette variante de chacun de leurs parents, mais c'est aussi le cas pour environ les deux tiers de l'ensemble de la population. Par conséquent, l'haplotype H1 semble être nécessaire, mais non suffisant pour engendrer une PSP.
Des recherches sont menées sur d'autres gènes, (éventuelles prédispositions génétiques), ainsi que sur les composants environnementaux toxiques comme sur d'autres contributeurs suspectés d'intervenir dans le déclenchement de la maladie.
Pour le syndrome de Guam et les syndromes parkinsoniens atypiques des Antilles dont la Guadeloupe, il a été mis en évidence, pour ces PSP, un risque de survenance supplémentaire par un facteur alimentaire (consommation notamment de corossol).
Traitement
Pour certains profils de PSP, et en début de maladie, les traitements adaptés à la maladie de Parkinson (dopamine, rivastigmine, etc.) peuvent être employés. Toutefois, en cas de résultats, ceux-ci demeurent souvent faibles et toujours temporaires. Par contre, il y a lieu de retenir que, pour le malade, tout gain pris, est pris.
Les traitements actuels, dans la durée, sont donc essentiellement symptomatiques et cherchent à lutter contre la gêne occasionnée par la maladie et son évolution. À ce titre la famille et l'entourage du patient sont essentiels, mais ils doivent eux-mêmes être soutenus et aidés sur le plan matériel et moral pour la mise en place de soins et traitements palliatifs en fonction de l'évolution de la maladie. Le fauteuil roulant et les aménagements induits, sont des équipements assez précoces dans la PSP.
Un règlement sur les médicaments orphelins a déjà été adopté par le parlement européen depuis le 15 décembre 1999.
Enfin, après avoir obtenu le statut de « médicament orphelin »[6] de la FDA et de l'EMEA (les deux agences des médicaments pour les États-Unis et l'Europe), deux médicaments, le Davunetide[7] et le ZentylorTM, sont en cours d'essai en phase II pour lutter contre la PSP. Ils ont obtenu l'accord de la FDA pour une procédure d'évaluation accélérée[8] dans le cadre des nouvelles dispositions prises sur les biotechnologies[9].
Depuis ce début d'année 2013, nous savons hélas, pour le Davunetide en essai de phase III, qu'aucun des critères d'évaluation secondaires ou exploratoires n'a montré des signes de changement. Il est à espérer que, malgré tout, cet échec apportera sa part de contribution positive à la lutte contre cette maladie.
Pronostic
La maladie n'est pas mortelle directement, par contre elle réduit l'espérance de vie de ceux qui en sont atteints. Compte tenu de l'errance de diagnostic d'une part et d'autre part de l'âge et de l'état de santé général par ailleurs du malade, cette espérance de vie pour un malade diagnostiqué PSP, dépasse rarement 15 ans. Les moyennes en la matière ne sont par contre pas significatives car elles ne reflètent pas les fortes disparités d'un malade à l'autre. La moyenne la plus admise est d'environ 7 ans. Les principales causes de réduction de l'espérance de vie, voire de mortalité, des malades PSP peuvent être :
- les chutes accidentelles en arrière, caractéristiques de la maladie ;
- les infections des voies respiratoires dues aux fausses routes répétées ;
- l'épuisement général et prématuré du malade, d'autant plus si le patient souffre d'autres pathologies et est âgé.
Recherches
Diagnostic
Examen neuropathologique post-mortem / Anatomopathologie
Comme pour les autres maladies neurodégénératives, il reste, à ce jour, le seul examen permettant de porter un diagnostic définitif de paralysie supranucléaire progressive. À cet égard, les patients et leur famille qui font don de leur cerveau[10], permettent des avancées considérables pour connaître et lutter contre la PSP et les maladies neurodégénératives.
S'il était établi qu'il y avait aussi une atteinte du système nerveux entérique chez les malades Parkinson[11], il semble exister une corrélation entre les lésions au système nerveux central et celles du système nerveux entérique. Cette avancée est considérable pour le dépistage et pour la mise au point et le « réglage » des médicaments actuels et à venir. En effet le système nerveux central n'est accessible qu'après le décès des malades. Ce n'est pas le cas des systèmes nerveux périphériques tel que le système nerveux entérique, ce qui présente l'avantage de pouvoir faire des investigations in vivo par une simple biopsie. La difficulté du diagnostic serait ainsi contournée.
Autres approches
Le projet européen NNIPPS, portant sur 800 patients s’est proposé en 2001 d’étudier les spécificités cliniques, cognitives, neuro-radiologiques et histologiques de la PSP. Les résultats définitifs publiés en 2011 ont hélas confirmé, que les essais n'avaient pas donné les résultats d'efficacité escomptée[12]. Des recherches sont également réalisées sur la nature des troubles oculomoteurs et de l’équilibre qui sont caractéristiques de l’affection.
Les maladies DCB, PSP, DFT dont la maladie de Pick sont considérées comme des taupathies (ou tauopathies), des maladies dues à des anomalies de la protéine tau, tout comme la maladie d'Alzheimer, plus connue en raison du nombre de patients concernés.
Toutefois la classification des pathologies neurodégénératives s'est compliquée depuis le milieu et la fin des années 1990.
Avec les approches moléculaire et génétique, pour les maladies neurodégénératives il est apparu une nouvelle classification et définition, basée sur la nature des constituants des lésions neuropathologiques :

- Les tauopathies regroupent des pathologies où les protéines microtubulaires tau sont impliquées. Des amas ou buissons de protéine tau se forment, s'accumulent et se propagent, entraînant de proche en proche la mort neurale des cellules contaminées. Les principales taupathies dégénératives de l’adulte sont la maladie d’Alzheimer, la paralysie supranucléaire progressive, la dégénérescence cortico-basale, et dont on rapproche aussi la maladie des grains argyrophiles, la maladie de Pick et les dégénérescences fronto-temporales avec syndrome parkinsonien liées au chromosome 17.
- Les synucléinopathies comportent des dépôts anormaux d'alpha-synucléines. Cette protéine pré-synaptique est un composant majeur des lésions de la maladie de Parkinson et des démences avec corps de Lewy, ainsi que des atrophies systémiques multiples. Dans les inclusions cytoplasmiques gliales de l'atrophie systématisée multiple, une accumulation de protéines alpha-synucléines est aussi observée.
La protéine FKBP52[13] agit comme une sorte de protéine anti-Tau dans la mesure où une importante présence de FKBP52 empêcherait l’accumulation de protéines Tau dans les neurones. La protéine FKBP52 est à ce titre en cours d'étude, avec un enjeu double : permettre un meilleur dépistage par la mesure de la quantité de protéine FKBP52 et élaborer des médicaments pour bloquer l'évolution de la pathologie tau.
Toutes ces expérimentations vont être poursuivies afin d'avoir des résultats plus larges de nature à modéliser des processus thérapeutiques, notamment basés sur des stratégies ciblant la protéine Tau[14].
En recherche appliquée des essais sont menés sur différentes molécules déjà connues mais dosées de sorte à pouvoir constituer un traitement contre la PSP. C'est le cas pour le lithium (États-Unis), Q10 (États-Unis et Royaume-Uni), valproate de sodium (France), chlorure de méthylthioninium, etc. À ce jour, même pour les molécules les plus prometteuses en premières phases d'essai, ces médicaments n'ont pas apporté les effets thérapeutiques escomptés.
Il y a lieu de citer aussi les essais[15] sur la protéine sphingomyélinase neutre (N-SMase)[16] qui, déclenchée par les lésions amyloïdes et DNF (où tau est impliquée), deviendrait inopérante dans le processus apoptotique de la mort neuronale, une fois cette protéine N-SMase inhibée. Le résultat escompté serait à la hauteur du capital neuronal ainsi préservé, notamment en termes de mémoire dans le cas de la maladie d’Alzheimer.
Mécanismes
La protéine tau, une protéine fortement impliquée
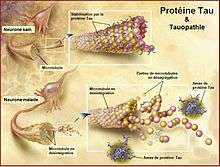
Le codage des protéines obéit à des mécanismes biochimiques et moléculaires complexes, à dessein d'assumer des fonctions normales de vie ou de mort (apoptose) de nos cellules.
Dans les années 1980, la dégénérescence neurofibrillaire est associée à la protéine tau qui, longtemps négligée par la recherche, suscite aujourd’hui un regain d’intérêt.
La protéine tau, comme toutes les autres protéines, est codée par nos gènes et le gène codant la protéine tau est situé sur le chromosome 17. Tout être humain fabrique de la protéine tau qui est normalement non pathogène et qui, même lorsqu'elle peut le devenir avec l'âge, demeure non toxique car sans expansion dans le système nerveux. Les mécanismes qui président au passage de la situation non pathogène de la protéine tau à pathogène, puis de pathogène à toxique, demeurent encore inexpliqués.
L'évolution de la maladie correspond à celle de tau dans le cerveau. La pathologie tau a une signature biochimique et morphologique de la dégénérescence neurofibrillaire. Par contre, il ne peut être espéré une relation très précise entre une région touchée et un signe cognitif précis, car d’autres facteurs aggravant interviennent.

Toutefois il faut retenir que la pathologie tau a un sens, dans la mesure où le chemin de la pathologie va dans le sens des signes cliniques. Par exemple pour Alzheimer : perte de mémoire (hippocampe touché) puis progressivement aphasie (cortex temporal médian et supérieur touché comme dans la démence sémantique, puis frontal antérieur et aire de Broca qui sont différentes aires du langage), apraxie (cortex frontal touché) agnosie (lobe pariétal, lobe occipital). L’atteinte frontale peut provoquer également des troubles du comportement.
Plus la pathologie tau avance dans le cortex, plus le chemin devient variable, selon la vulnérabilité individuelle. La variation de l’atteinte des régions du cortex associatif puis primaire provoque les variations cliniques inter-individuelles marquées. C’est pour cela que chaque maladie neurodégénérative a un profil général et statistique assez précis et constant, mais que chaque cas a sa spécificité. Ce profil d'évolution peut être représenté, par exemple pour la maladie d’Alzheimer, en 10 stades caractéristiques d'atteintes.
Organisation du traitement, des soins et de la recherche en France

Depuis mai 2007, dans le cadre du premier Plan National Maladies Rares[19], en liaison avec l'association PSP France, un centre de référence a été labellisé pour la paralysie supranucléaire progressive au sein de l'hôpital de la Salpétrière à Paris.
Depuis mars 2009, le réseau national des centres de référence et de compétence pour la PSP[20] vise à apporter un supplément d'expertise dans la prise en charge, le traitement et la recherche. L'association PSP France (pour les PSP et DCB), rejointe début 2010 par l’association France-DFT (pour les DFT) sont à ce jour les deux seules associations de malades présentes au sein de ce réseau pour les démences rares, au sens médical du terme et non pas psychiatrique (classement nosologique).[pas clair]
Depuis le , le PNMR (Plan national Maladies rares) a été reconduit et doté d'un budget de 180 millions d'euros[21].
La recherche, dont le centre NeuroSpin, vise à mieux comprendre[22] les mécanismes de la dégénérescence neuronale, pour mieux les reconnaître et les prévenir ; pour mieux ralentir l’évolution de ce type de maladie, voire la guérir.
Personnalités atteintes de la maladie
- Peter Sarstedt (1941-2017), chanteur anglais[23].
- Abdus Salam (1926-1996), physicien pakistanais récipiendaire du prix Nobel de physique 1979 avec Sheldon Glashow et Steven Weinberg.
Notes et références
- Charles Dickens (1857), littérature et PSP
- (en) Richardson JC, Steele J, Olszewski J, « Supranuclear ophthalmoplegia, pseudobulbar palsy, nuchal dystonia and dementia. A clinical report on eight cases of 'heterogeneous system degeneration' », Transactions of the American Neurological Association, vol. 88, , p. 25–9 (PMID 14272249)
- (en) I. Litvan et al. « Accuracy of the clinical diagnosis of corticobasal degeneration: a clinicopathologic study » Neurology 1997: 48: 119-25.
- (en) Samii A, Nutt JG, Ransom BR, « Parkinson's disease », Lancet, vol. 363, no 9423, , p. 1783–93 (PMID 15172778, DOI 10.1016/S0140-6736(04)16305-8)
- (en) Lesage S, Brice A, « Parkinson's disease: from monogenic forms to genetic susceptibility factors », Hum. Mol. Genet., vol. 18, no R1, , R48–59 (PMID 19297401, DOI 10.1093/hmg/ddp012)
- [PDF] Médicament Orphelin
- L'étude multi-nationale du Davunetide, qui porte sur 300 patients, est menée dans les plus importantes institutions médicales des États-Unis, d'Australie, d'Allemagne, de France, du Canada et du Royaume-Uni.
- [PDF] Procédure accélérée
- [PDF] Dossier des biotechnologies
- Site explicatif pour le don de cerveau
- [PDF] PLoS ONE, September 2010 | Volume 5 | Issue 9 | e12728
- [PDF] PLoS ONE, August 2011 | Volume 6 | Issue 8 | e22293
- [PDF] cpINSERM 26-janv-2010
- [PDF]Stratégie ciblant la protéine Tau, Prise en charge de la maladie d'Alzheimer : données actuelles et en cours de développement - Mars 2011 Extraits
- [PDF] Nouvelle cible contre Alzheimer
- Annales de l'Institut Pasteur / Actualités - Volume 11, Issue 2, 6 April 2000, Pages 25-45.
- (en) Delacourte A, David JP, Sergeant N et al. « The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer's disease » Neurology 1999;52:1158-65.
- (en) A Delacourte A et L Buée « Normal and pathological Tau proteins as factors for microtubule assembly » Int Rev Cytol. 1997;171:167-224.
- [PDF] Plan National Maladies Rares 2005-2008
- Réseau National des Centres de Référence et de Compétence pour la PSP
- Plan National Maladies Rares 2010-2014
- [PDF] Neurospin 24 novembre 2006
- «Singer-songwriter Peter Sarstaedt dies aged 75», BBC, 8 janvier 2017
{kind=link}
Références externes
- Le cerveau à tous les niveaux : Un blogue canadien à consulter.
- Réseau Centres de Référence et Compétence des Démences Rares
- ORPHANET, le portail des maladies rares et des médicaments orphelins
- PSP 39 - Parkinson Syndromes Parkinsoniens
- Fondation pour la PSP - États-Unis
- Association Paralysie Supranucléaire Progressive - PSP France
Galerie d'images
 Mésencéphale et rhombencéphale (vue postero-latérale).
Mésencéphale et rhombencéphale (vue postero-latérale). Base du cerveau.
Base du cerveau.
 De l'œil au cortex visuel.
De l'œil au cortex visuel. Schéma des voies optiques entre les deux rétines et le cortex visuel.
Schéma des voies optiques entre les deux rétines et le cortex visuel.
