Syndrome POEMS
Le syndrome POEMS (en anglais : POEMS syndrome) est une association de symptômes dont le nom est un acronyme anglais décrivant les principaux éléments constitutifs du syndrome :
- « P »olyneuropathy : Polyneuropathie périphérique, atteinte du système nerveux périphérique.
- « O »rganomegaly : Organomégalie, agrandissement anormal des organes.
- « E »ndocrinopathy : Endocrinopathie, maladies endocriniennes.
- « M »onoclonal protein : Dysglobulinémie monoclonale, traduite par la présence d'une immunoglobuline monoclonale en excès.
- « S »kin changes : Anomalie cutanée
Manifestations du syndrome
Le syndrome n'est pas toujours complet et certains de ses symptômes peuvent manquer, à l'inverse d'autres symptômes peuvent être présents tel que : fatigue, douleur, perte de poids, fébricule, papillœdème, œdème, épanchement pleural, ascite, anasarque, hypertension pulmonaire primitive, diarrhée, insuffisance rénale, lésions osseuses cancéreuses (plasmocytome), maladie de Castelman et au niveau biologique thrombocytose et polyglobulie.
La cause de ce syndrome n'est pas encore connue même si de nombreuses hypothèses physiopathologiques existent.
Cette maladie est exceptionnelle, sa fréquence exacte n'est pas connue, elle touche plus les hommes que les femmes et l'âge moyen de survenue se situe autour de 50-60 ans avec des extrêmes allant de 25 à 83 ans. Le POEMS syndrome pose un problème diagnostique, car c'est une maladie rare donc peu connue des médecins qui diagnostiquent parfois à tort une polyneuropathie inflammatoire chronique démyélinisante (CIDP) car le symptôme principal est une polyneuropathie périphérique ayant les caractéristiques d'une CIDP. Ceci est dommageable car les traitements efficaces pour un CIDP (gammaglobuline et plasmaphérèse) ne le sont pas dans le POEMS. Or une prise en charge précoce de la maladie est essentielle, car elle permet d'en réduire la morbidité (diminution des séquelles neurologiques), d'où la nécessité d'un diagnostic précis et rapide.
Les clés pour aboutir au bon diagnostic sont, outre les éléments cliniques, la présence d'une thrombocytose à la prise de sang et de lésions osseuses évocatrices sur les radiographies corps entier.
Le POEMS est une maladie grave et la réponse aux traitements est le facteur pronostic essentiel.
La prise en charge dépend de l'extension de la maladie. Si elle est peu développée, la chirurgie et l'ablation du plasmocytome puis la radiothérapie sont préconisées. Si la maladie est plus diffuse on propose une corticothérapie, une chimiothérapie à base d’alkylants voir une autogreffe de cellules souches hématopoïétiques après une chimiothérapie intensive.
Synonymes
Le syndrome POEMS est également dénommé[1]:
- Crow-Fukase syndrome ou syndrome de Crow-Fukase ; dénomination principalement utilisée au Japon[2] ;
- Takatsuki syndrome ou syndrome de Takatsuki[3] ;
- PEP syndrome : polyneuropathy endocrinopathy plasma cell dyscrasia ;
- polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, skin changes ;
- Shimpo syndrome[4]
Classification internationale des maladies (CIM)
- CIM 10 :
Historique
Les associations entre les dysglobulinémies les neuropathies périphériques sont bien connues depuis longtemps[5]. De un tiers à la moitié des patients ayant un myélome ostéocondensant ont une neuropathie[6],[7], et la moitié des patients ayant un myélome et une neuropathie périphérique ont des lésions ostéocondensantes. Ces chiffres contrastent fortement avec les 1 à 8 % de neuropathies retrouvées chez des patients avec un myélome classique[8],[9].
En 1956, ce lien entre les dysglobulinémie et les neuropathies périphériques fut démontré par Crow[5], qui publia la description de 2 patients associant plasmocytome ostéocondensant, neuropathie et autres symptômes tels que hippocratisme digital, zone d'hyperpigmentation ou de décoloration cutanée, ongle blanc, adénopathie et œdème.
En 1938, Scheinker[10] publia une étude autopsique qui peut être considérée comme la première description de POEMS syndrome. Le patient de 39 ans avait un plasmocytome solitaire, une polyneuropathie sensitivo-motrice et des zones cutanées épaissies et hyperpigmentées sur la poitrine[11].
Par la suite il y eut de nombreux autres reports de patients associant myélome ostéocondensant et neuropathie périphérique avec organomégalie, anomalie cutanée, endocrinopathie, œdème, hypertrichose, gynécomastie et ascite[7],[6],[11],[12],[13],[14],[15],[16],[17],[18],[19],[20].
En 1977, Iwashita et al.[11] publia une étude comparant 30 patients avec un myélome ostéocondensant et une neuropathie périphérique et 29 patients avec un myélome sans neuropathie périphérique. Le premier groupe avait un taux plus élevé d'hyperpigmentation, d'hypertrichose, d'œdème, d'épaississement cutanée, d'hépatomégalie et d'hippocratisme digital.
En 1980, Bardwick et al.[21] créa l'acronyme POEMS pour désigner l'association des symptômes polyneuropathy, organomegaly, endocrinopathy, M protein, et skin changes. De nombreux autres symptômes importants ne figurent pas dans cet acronyme tels que les lésions osseuses condensantes, la maladie de Castleman, l'œdème papillaire, l'épanchement pleural, œdème, ascite et thrombocytose[22],[23],[24],[25].
Épidémiologie
Le POEMS syndrome est une maladie rare, sa prévalence exacte reste inconnue.
Plus de 300 cas ont été décrits dans la littérature, les principales séries sont toutes rétrospectives :
- en 1984 la série historique japonaise de 102 cas[22],
- en 1992 une série de la Mayo Clinic avec 39 patients[26],
- en 1994 une série multicentrique française de 25 cas[24],
- en 1998 la série la plus récente de la Mayo Clinic regroupant 99 patients[27] étudiés sur une période allant de 1975 à 1998.
L'homme est plus souvent touché, avec un sex ratio approchant les 2/3 pour l'homme. Ainsi 63 % des patients sont de sexe masculin dans la série de la Mayo Clinic de 1998.
L'âge de début du POEMS est plus précoce que pour le myélome. Le pic d'incidence du syndrome POEMS se situe entre la 5e et la 6e décennie[24],[27], avec des extrêmes allant de 25[28] à 83 ans[27]. L'âge moyen de survenue varie selon les séries, il est de 51 ans dans les 2 séries de la Mayo Clinic. Et dans celle de 1998, 31 % des patients étaient âgés de 45 ans ou moins.
Physiopathologie
La physiopathologie n'est pas encore très claire. Différentes hypothèses ont été avancées mais aucune n'a été réellement confirmée.
- Pathogénie directe de la chaîne légère lambda : la responsabilité de la dysglobulinémie monoclonale dans la genèse de la symptomatologie clinique est suggérée par la disparition de celle-ci lors des thérapeutiques efficaces de la dyscrasie plasmocytaire. Mais l’absence d’immunoglobuline monoclonale sérique dans de nombreuses observations plaide contre cette théorie. Et si une chaîne légère lambda est retrouvée dans plus de 95 % des cas, les études histopathologiques des différents organes et nerfs atteints ne retrouvent pas signe évoquant une anomalie due à des dépôts.
- Anomalie génétique : Soubrier et al. ont décelé en 2004 des anomalies au niveau des gènes Vlambda chez 2 patients atteints de POEMS syndrome[29].
- Responsabilité de l'HHV 8 : des anticorps contre le virus herpès humain (HHV) 8 ont été trouvés chez 78 % des patients ayant un poems et une maladie de Castelman, et chez seulement 22 % de ceux qui ont un poems sans Castelman[30].
- Cytokines et VEGF, hypothèse vasculaire : on trouve également des taux élevés de cytokines, et en particulier de VEGF, qui semble jouer un rôle dans la physiopathologie du poems[31],[32],[33],[34],[35].
Le VEGF entraîne une augmentation de la perméabilité vasculaire et joue un rôle important dans l'angiogénèse. Il est normalement présent dans les ostéoblastes et il pourrait être un régulateur important dans la différenciation ostéoblastique. Une théorie serait que le VEGF soit fabriqué dans les cellules plasmatiques et les plaquettes, entraînant une augmentation de la perméabilité vasculaire, de l'angiogénèse et de la migration des monocytes et macrophages, ce qui pourrait entraîner potentiellement des obstructions vasculaires. Hashiguchi a montré que chez des patients atteints de POEMS, les agrégats plaquettaires relâchaient du VEGF[35].
Le VEGF pourrait être responsable de l'organomégalie, des œdèmes et des lésions cutanées. Son rôle dans la neuropathie est moins évident, mais il pourrait être responsable de microthromboses dans les vaisseaux endoneuronaux. Watanabe et al. (1998)[36] avancent que la surproduction de VEGF pourrait expliquer la microangiopathie, la néovascularisation et l'augmentation de la perméabilité vasculaire. Ils ont trouvé que le taux sérique de VEGF, chez 10 patients atteints de POEMS, était 15 à 30 fois plus élevé que chez des patients atteints de Guillain-Barré, d'une polyneuropathie inflammatoire chronique démyélinisante (CIDP), et d'autres atteintes neurologiques. Le taux de VEGF dans le LCR était par contre similaire.
- Le système MMP/TIMP : des taux élevés de métalloprotéases matricielles (MMP) et de leurs inhibiteurs physiologiques, les TIMP (tissue inhibitors of metalloproteinases) ont été observés chez des patients atteints de POEMS. Les taux sériques de VEGF et de TIMP-1 semblent fortement corrélés mais on ne comprend pas encore la signification de ces observations[38].
Clinique
- La neuropathie est constante, souvent révélatrice, sensitivo-motrice et symétrique.
Les nerfs crâniens, et le système nerveux autonome (dysautonomie) sont épargnés.
L'examen du LCR montre une hyperprotéinorachie sans réaction cellulaire.
Le résultat de la biopsie neuro-musculaire n'est pas spécifique (pas d'amylose, pas de dépôt d'immunoglobuline, pas d'infiltration plasmocytaire).
Toujours dans le cadre de cette neuropathie, un œdème papillaire au fond d'œil peut se voir.
- L'organomégalie concerne la rate, le foie, et les ganglions lymphatiques.
L'histologie hépatique est en général normale, en l'absence de pathologie associée toujours possible.
Une maladie de Castelman peut être décelée à la biopsie ganglionnaire (hyperplasie angiofolliculaire). La plupart du temps il n'existe qu'une adénite non spécifique.
- L'endocrinopathie concerne diversement :
- les gonades : hypoandrie, gynécomastie, aménorrhée, hyperprolactinémie, hyperœstrogénie ;
- la thyroïde : hypothyroïdie ;
- un diabète, peu intense.
- Les anomalies cutanées concernent une hypertrichose, un épaississement de la peau, une hyperpigmentation et des angiomes.
Il peut y avoir aussi de l'œdème, (mais aussi plus rarement une ascite et des épanchements pleuraux). Un phénomène de Raynaud a été décrit ainsi qu'une hypertension artérielle pulmonaire et des thromboses aussi bien veineuses qu'artérielles.
Diagnostic
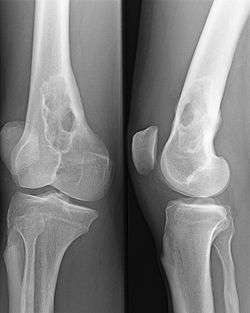
- Il existe des anomalies biologiques :
- thrombocytose ;
- augmentation de la vitesse de sédimentation ;
- polyglobulie et/ou hyperleucocytose ;
- plus rarement détérioration biologique de la fonction rénale.
- Il existe une dyscrasie plasmocytaire, terme volontairement imprécis qui ne fait pas référence au caractère malin ou non de cette prolifération. Le myélogramme et la biopsie de moelle peuvent être normaux ou montrer une infiltration plasmocytaire ou lympho-plamocytaire.
- La conséquence en est la dysglobulinémie monoclonale, marquée par un pic dans les globulines à l'électrophorèse, confirmée à l'immuno-électrophorèse, et au dosage des immunoglobulines. Il s'agit surtout d'IgG ou d'IgA.
- Le bilan osseux (Radiographies, IRM, scintigraphie osseuse) peut montrer des lésions multiples condensantes, évocatrices d'un myélome multiple, ou d'un plasmocytome solitaire, dont le diagnostic et le traitement sera chirurgical.
- Le diagnostic différentiel est protéiforme, selon la prédominance de la symptomatologie clinique. Peuvent ainsi pouvoir être discutés :
- la sclérodermie ;
- un diabète avec neuropathie spécifique ;
- le diagnostic différentiel entre le myélome multiple (maladie de Kahler), et le POEMS syndrome est plus nosologique que pratique.
Le diagnostic est particulièrement difficile dans les pays où certains examens comme les analyses immunologiques, l’EMG et le TDM sont difficilement réalisables pour des raisons d’accessibilité économique[39].
Critères diagnostiques
En 2007 des critères diagnostiques sont proposés[40]. Tous les critères suivants doivent être présents pour que le diagnostic soit posé :
- polyneuropathie
- prolifération monoclonale
- un ou plusieurs des critères majeurs suivants :
- lésions osseuses ;
- maladie de Castleman ;
- augmentation des taux plasmatiques de VEGF.
- un ou plusieurs des critères mineurs suivants :
- organomégalie (augmentation de volume de la rate, du foie, ou des ganglions lymphatiques) ;
- surcharge du compartiment extra-vasculaire (œdème, pleurésie, ascite) ;
- endocrinopathie, le diabète ou l'hypothyroïdie seuls sont insuffisants ;
- signes cutanés ;
- œdème papillaire ;
- thrombocytose ou polycythémie.
Traitement et prise en charge
Lorsqu'il existe un plasmocytome solitaire, le traitement de choix est chirurgical. Il peut bénéficier d'une radiothérapie, pré ou post opératoire.
Les lésions osseuses diffuses bénéficient d'un protocole chimiothérapique comparable à celui d'un myélome. Les résultats sont inconstants.
Dans les formes suffisamment sévères pour mettre en jeu à court terme le pronostic vital, une chimiothérapie à haute dose suivie d'une greffe de moelle peut être proposée.
Les échanges plasmatiques (Plasmaphérèse) sont en théorie susceptibles d'améliorer les neuropathies sévères, mais inconstamment et transitoirement.
Évolution, complication, pronostic
- Le syndrome est d'évolution lente, pouvant dépasser la décennie.
- La polyneuropathie peut aboutir à une paralysie avec état de dépendance grabataire.
- La dysglobulinémie peut se compliquer d'anomalies rénales.
- Une insuffisance cardiaque terminale est possible, dont la physiopathologie est complexe.
L'évolution se fait sur un mode chronique. Le pronostic a souvent été décrit comme pauvre, avec des médiane de survie faibles allant de 12 à 33 mois[6],[22],[12].
Pourtant lors de la dernière étude de la Mayo Clinic publiée en 2003, la médiane de survie retrouvée chez les patients traités sans autogreffe de cellule souche était de 165 mois (13.8 ans)[27].
Les études retrouvant des patients ayant la maladie depuis plus de 5 ans ne sont pas rares[9],[19],[41],[42],[43],[44].
Dans une étude, 7 patients sur 15 vivaient encore après plus de 5 ans, et un patient vivait avec la maladie depuis 25 ans[45].
Les données de la littérature montrent que la survie n'a aucun lien avec le nombre de symptômes retrouvés[24]. Il semblerait que l'hippocratisme digital et les œdèmes soient associés à une survie plus courte, la médiane étant dans ces 2 cas respectivement de 31 et 79 mois[27].
Dans la série de la Mayo Clinic étudiant 99 patients atteints de POEMS de 1975 à 1998, 35 étaient décédés[27]. Les causes de décès les plus fréquentes étaient l'insuffisance cardiorespiratoire, et les infections. Aucun patient, même ceux ayant des lésions osseuses multiples n'avait développé de myélome multiple. Les fractures osseuses étaient rares. Les patients ayant été traités par radiothérapie et ceux ayant présenté une très bonne ou bonne réponse au traitement présentaient une meilleure survie.
Associations de patients
Notes et références
- Syndrome POEMS sur http://www.chu-rouen.fr
- Lagueny et al. Le syndrome POEMS (ou syndrome de Crow-Fukase) Revue neurologique, 2004, vol. 160, no3, pp. 285-295.
- (en) Pruzanski W. Takatsuki syndrome: a reversible multisystem plasma cell dyscrasia. Arthritis Rheum 1986 Dec; 29(12):1534-5.
- (en) Shimpo S 1968. Solitary myeloma causing polyneuritis and endocrine disorders. Nippon Rinsho, 26: 2444-2456.
- (en) Crow R. « Peripheral neuritis in myelomatosis » Brit Med J. 1956;2:802-804. PMCID: PMC2035359
- (en) Driedger H, Pruzanski W. « Plasma cell neoplasia with osteosclerotic lesions: a study of five cases and a review of the literature » Arch Intern Med. 1979;139:892-896. Abstract
- (en) Mangalik A, Veliath AJ. « Osteosclerotic myeloma and peripheral neuropathy: a case report » Cancer 1971;28:1040-1045. Abstract
- (en) Evison G, Evans KT. « Sclerotic bone deposits in multiple myeloma [letter] » Br J Radiol. 1983;56:145.
- Reitan JB, Pape E, Fossa SD, Julsrud OJ, Slettnes ON, Solheim OP. « Osteosclerotic myeloma with polyneuropathy » Acta Med Scand. 1980;208:137-144.
- (de) Scheinker I. « Myelom und Nervensystem: Über eine bisher nicht beschriebene mit eigentümlichen Hautveränderungen einhergehende Polyneuritis bei einem plasmazellulären Myelom des Sternums » Deutsche Zeitschrift für Nervenheilkunde 1938;147:247-273. springerlink
- (en) Iwashita H, Ohnishi A, Asada M, Kanazawa Y, Kuroiwa Y. « Polyneuropathy, skin hyperpigmentation, edema, and hypertrichosis in localized osteosclerotic myeloma » Neurology 1977;27:675-681. Abstract
- (en) Driedger H, Pruzanski W. « Plasma cell neoplasia with peripheral polyneuropathy: a study of five cases and a review of the literature » Medicine 1980;59:301-310. Abstract
- (en) Morley JB, Schwieger AC. « The relation between chronic polyneuropathy and osteosclerotic myeloma » J Neurol Neurosurg Psychiatry 1967;30:432-442. Abstract
- (ja) Shimpo S. « Solitary myeloma causing polyneuritis and endocrine disorders [in Japanese] » Nippon Rinsho. 1968;26:2444-2456. Abstract
- (en) Mayo CM, Daniels A, Barron KD. « Polyneuropathy in the osteosclerotic form of multiple myeloma » Trans Am Neurol Assoc. 1968;93:240-242. Abstract
- (en) Fukase M, Kakimatsu T, Nishitani H et al. « Report of a case of solitary plasmacytoma in the abdomen presenting with polyneuropathy and endocrinological disorders » Clin Neurol. 1969;9:657.
- (en) Imawari M, Akatsuka N, Ishibashi M, Beppu H, Suzuki H. « Syndrome of plasma cell dyscrasia, polyneuropathy, and endocrine disturbances: report of a case » Ann Intern Med. 1974;81:490-493. Abstract
- (en) Getaz P, Handler L, Jacobs P, Tunley I. « Osteosclerotic myeloma with peripheral neuropathy » S Afr Med J. 1974;48:1246-1250. Abstract
- (en) Waldenström JG, Adner A, Gydell K, Zettervall O. « Osteosclerotic "plasmocytoma" with polyneuropathy, hypertrichosis and diabetes » Acta Med Scand. 1978;203:297-303. Abstract
- (en) Moya-Mir MS, Martin-Martin F, Barbadillo R et al. « Plasma cell dyscrasia with polyneuritis and dermato-endocrine alterations: report of a new case outside Japan » Postgrad Med J. 1980;56:427-430. Abstract
- (en) Bardwick PA, Zvaifler NJ, Gill GN, Newman D, Greenway GD, Resnick DL. « Plasma cell dyscrasia with polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes: the POEMS syndrome: report on two cases and a review of the literature » Medicine 1980;59:311-322. Abstract
- (en) Nakanishi T, Sobue I, Toyokura Y et al. « The Crow-Fukase syndrome: a study of 102 cases in Japan » Neurology 1984;34:712-720. Abstract
- (en) Milanov I, Georgiev D. « Polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin changes (POEMS) syndrome » Can J Neurol Sci. 1994;21:60-63. Medline
- (en) Soubrier MJ, Dubost JJ, Sauvezie BJ. « POEMS syndrome: a study of 25 cases and a review of the literature. French Study Group on POEMS syndrome » Am. J. Med. 1994;97:543-553. Medline
- (en) Longo G, Emilia G, Torelli U. « Skin changes in POEMS syndrome » Haematologica 1999;84:86. Abstract
- (en) Miralles GD, O'Fallon JR, Talley NJ. « Plasma-cell dyscrasia with polyneuropathy: the spectrum of POEMS syndrome » N Engl J Med. 1992;327:1919-1923. Abstract
- (en) Dispenzieri A, Kyle RA, Lacy MQ et al. « POEMS syndrome: definitions and long-term outcome » Blood 2003;101:2496–2506. Full Text
- (en) Wong VA, Wade NK. « POEMS syndrome: an unusual cause of bilateral optic disk swelling » Am J Ophthalmol. 1998;126:452–454
- (en) Martin Soubrier, Labauge P, Jouanel P, Viallard JL, Piette JC, Sauvezie B. « Restricted use of Vlambda genes in POEMS syndrome » Haematologica
- (en) Belec L, Authier FJ, Mohamed AS, Soubrier M, Gherardi RK. « Antibodies to human herpesvirus 8 in POEMS (polyneuropathy, organomegaly, endocrinopathy, M protein, skin changes) syndrome with multicentric Castleman’s disease » Clin Infect Dis. 1999;28:678–679
- (en) Gherardi RK, Belec L, Soubrier M, et al. « Overproduction of proinflammatory cytokines imbalanced by their antagonists in POEMS syndrome » Blood 1996;87:1458–1465.
- (en) Hitoshi S, Suzuki K, Sakuta M. « Elevated serum interleukin-6 in POEMS syndrome reflects the activity of the disease » Intern Med. 1994;33:583–587.
- (en) Rose C, Zandecki M, Copin MC et al. « POEMS syndrome: report on six patients with unusual clinical signs, elevated levels of cytokines, macrophage involvement and chromosomal aberrations of bone marrow plasma cells » Leukemia 1997;11:1318–1323.
- (en) Feinberg L, Temple D, de Marchena E, Patarca R, Mitrani A. « Soluble immune mediators in POEMS syndrome with pulmonary hypertension: case report and review of the literature » Crit Rev Oncog. 1999;10:293–302.
- (en) Hashiguchi T, Arimura K, Matsumuro K et al. « Highly concentrated vascular endothelial growth factor in platelets in Crow-Fukase syndrome » Muscle Nerve 2000;23:1051–1056.
- (en) Watanabe O, Maruyama I, Arimura K et al. « Overproduction of vascular endothelial growth factor/vascular permeability factor is causative in Crow-Fukase (POEMS) syndrome » Muscle Nerve 1998;21:1390–1397.
- (en) Soubrier M, Dubost JJ, Serre AF et al. « Growth factors in POEMS syndrome: evidence for a marked increase in circulating vascular endothelial growth factor » Arthritis Rheum. 1997;40:786–787.
- (en) Michizono K, Umehara F, Hashiguchi T et al. « Circulating levels of MMP-1, -2, -3, -9, and TIMP-1 are increased in POEMS syndrome » Neurology 2001;56:807–810.
- (en) « Neuropathie périphérique révélatrice d'un POEMS syndrome : à propos d'un cas - POEMS syndrome revealed by a peripheral neuropathy » African journal of neurological sciences 2005, vol 24, no 2.
- (en) Dispenzieri, A. (2007) « POEMS syndrome » Blood reviews 21 (6): 285–299 DOI:10.1016/j.blre.2007.07.004
- (en) Sakemi H, Okada H. « An autopsy case of Crow-Fukase syndrome which developed 18 years after the first manifestation of plasmacytoma » Intern Med. 1992;31:50-54.
- (en) Cabezas-Agricola JM, Lado-Abeal JJ, Otero- Anton E, Sanchez-Leira J, Cabezas-Cerrato J. « Hypoparathyroidism in POEMS syndrome » Lancet 1996;347:701-702
- (en) Rongioletti F, Gambini C, Lerza R. « Glomeruloid hemangioma: a cutaneous marker of POEMS syndrome » Am J Dermatopathol. 1994;16:175-178
- (en) Silberstein LE, Duggan D, Berkman EM. « Therapeutic trial of plasma exchange in osteosclerotic myeloma associated with the POEMS syndrome » J Clin Apheresis. 1985;2:253-257.
- (en) Gherardi RK, Belec L, Soubrier M et al. « Overproduction of proinflammatory cytokines imbalanced by their antagonists in POEMS syndrome » Blood 1996;87:1458-1465. Abstact.
Liens externes
- Hematology 2005 © 2005 The American Society of Hematology - POEMS Syndrome - Angela Dispenzieri
- Blood. 2003 Apr 1;101(7):2496-506. Epub 2002 Nov 27. - POEMS syndrome: definitions and long-term outcome. - Dispenzieri A et al. - Division of Hematology and Internal Medicine, Mayo Clinic, Rochester, MN 55905, États-Unis
- Résumé orphanet - ORPHA2905 et fiche complète PDF : Angela Dispenzieri et Morie A.Gertz, mars 2005.
- POEMS syndrome: mise au point - Dr A. Nouijai janvier 2005
- CRI : iconographie diverse
- POEMS Syndrome - Dr Joanna L. Chan -emedicine

