Cholestérol
Le cholestérol est un lipide de la famille des stérols qui joue un rôle central dans de nombreux processus biochimiques. Le cholestérol tire son nom du grec ancien chole- (bile) et de stereos (solide), car il fut découvert sous forme solide dans les calculs biliaires en 1758 par François Poulletier de La Salle. Mais ce n'est qu'en 1814 que le chimiste français Eugène Chevreul lui donna le nom de cholestérine.
| Cholestérol | |
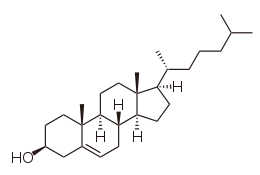 | |
| Structure du cholestérol. | |
| Identification | |
|---|---|
| Nom UICPA | (3S,8S,9S,10R,13R,14S,17R)-10,13-dimethyl-17-[(2R)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-ol |
| No CAS | |
| No ECHA | 100.000.321 |
| No EC | 200-353-2 |
| DrugBank | DB04540 |
| PubChem | |
| SMILES | |
| InChI | |
| Apparence | solide blanc |
| Propriétés chimiques | |
| Formule brute | C27H46O [Isomères] |
| Masse molaire[1] | 386,6535 ± 0,0251 g/mol C 83,87 %, H 11,99 %, O 4,14 %, |
| Propriétés physiques | |
| T° fusion | 147 à 150 °C[2] |
| T° ébullition | 360 °C (décomposition)[2] |
| Solubilité | pratiquement insoluble (eau)[2] |
| Masse volumique | 1,07 g·cm-3 (20 °C)[2] |
| Point d’éclair | 250 °C[2] |
| Cristallographie | |
| Classe cristalline ou groupe d’espace | P1[3] |
| Paramètres de maille | a = 27,565 Å b = 38,624 Å |
| Volume | 10 151,15 Å3[3] |
| Précautions | |
| Classification du CIRC | |
| Groupe 3 : inclassable quant à sa cancérogénicité pour l'homme[4] | |
| Unités du SI et CNTP, sauf indication contraire. | |
Le mot « cholestérol » désigne une molécule unique. Ce qui signifie que les termes de « bon » et « mauvais cholestérol » ne servent pas à désigner deux molécules différentes, mais font référence aux lipoprotéines de haute densité (HDL) et lipoprotéines de basse densité (LDL), les transporteurs du cholestérol dans le sang. Voir en particulier la teneur en cholestérol dans l'alimentation et athérosclérose.
Histoire
Le cholestérol est découvert sous forme solide dans les calculs biliaires en 1758 par François Poulletier de La Salle[5].
Alors que les scientifiques pensaient que les maladies cardiovasculaires étaient principalement la conséquence de l'âge, l'étude de Framingham qui a débuté depuis 1948, montre l'importance d'autres facteurs de risque[6] : le tabac, le diabète, l'hypertension artérielle, un régime alimentaire riche en cholestérol (cette étude suggère en 1961[7] le rôle du LDL). En 1976 est élaboré à partir de cette étude un « score de risques » (amélioré en 1998) permettant de détecter les personnes les plus susceptibles de déclencher une maladie cardio-vasculaire en fonction de ces différents éléments[8].
Le nutritionniste américain Ancel Keys réalise après la Seconde Guerre mondiale l'étude des 7 pays (en) (ne prenant pas en compte la France et la Finlande qui ne valident pas la courbe présentée pour ces 7 pays)[9], étude épidémiologique sur plusieurs décennies qui met en évidence une corrélation[10] entre le taux de cholestérol sanguin et les accidents cardiovasculaires. Ces résultats lui font émettre l'« hypothèse lipidique » selon laquelle le cholestérol est le facteur de risque majeur responsable de la forte mortalité cardio-vasculaire mais cette étude souffre de biais de comparaison ou des confondeurs[11].
À la suite de cette étude longitudinale, des essais cliniques sont menés sur des populations d'anciens combattants américains mis au régime hypocholestérolémiant mais ces tentatives n'ont pas d’impact significatif sur leur mortalité, l'« hypothèse lipidique » n'est pas validée[12].
En 1954, le chercheur français Jean Cottet découvre que des ouvriers agricoles intoxiqués par le pesticide qu'ils répandent dans les champs ont un taux de cholestérol qui s'est effondré. Un de ses amis chimiste de l'Imperial Chemical Industries (Michael Oliver) synthétise un médicament dérivé de ce pesticide, le clofibrate. Le test de cette molécule sur des rats puis des patients confirme son effet hypolipémiant[13]. L'Organisation mondiale de la santé réalise un essai clinique sur 15 000 Européens pour évaluer l'effet du clofibrate sur la prévention de l'infarctus, mais cette étude est négative, l'essai devant même être arrêté prématurément, le groupe sous clofibrate ayant une prévalence plus élevée que le groupe sous placebo[14].
Malgré cette étude réfutant le lien entre baisse du cholestérol et surmortalité, une famille de molécules médicamenteuses hypolipémiantes dérivées de ce médicament est lancée, les fibrates.
Au milieu des années 1960, l'industrie sucrière nord-américaine, à travers son syndicat professionnel, la Sugar Research Foundation (en), finance des chercheurs de Harvard pour discréditer la validité des études scientifiques faisant la relation entre un régime riche en sucre et les maladies cardiovasculaires (une glycémie élevée due à l'ingestion de sucres rapides provoque un stress oxydatif et inflammatoire sur les parois artérielles)[15].
En 1969, le chercheur Kilmer S. McCully (en) observe des taux élevés d'homocystéine dans le sang des sujets atteints d'affections cardiovasculaire. Récusant l'« hypothèse lipidique », il pense que cet acide aminé joue un rôle dans l'athérosclérose, ce qui lui vaut de perdre son poste de la Harvard Medical School and Massachusetts General Hospital[16].
En 1973, le biochimiste Akira Endō découvre la première statine. Dans les années 1990, deux études sur la simvastatine[17] et la pravastatine[18] montrent leur effet de prévention sur des hommes ayant un taux de cholestérol élevé. Certains[19],[20] soulignent que les recommandations[21] tendant à viser un taux optimal de cholestérol (plus particulièrement sa fraction LDL), ne sont en fait étayées par aucune étude, ces dernières ayant toujours été faites à des doses fixes de statines quel que soit le taux initial de cholestérol, la baisse de ce dernier n'étant pas un objectif[22]. Il semble que les statines ont un effet de prévention vasculaire mais sans lien avec le taux de cholestérol[23].
Structure
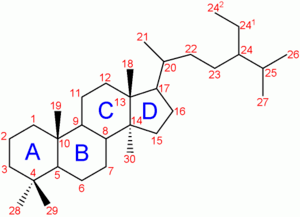
La molécule de cholestérol comprend quatre cycles carbonés notés A, B, C et D (noyau cyclopentano-perhydro-phénanthrénique), 8 carbones asymétriques (les carbones 3, 8, 9, 10, 13, 14, 17 et 20), ce qui fait 28 soit 256 stéréoisomères dont un seul existe : le 3β-ol lévogyre. Le cholestérol possède un groupe hydroxyle -OH sur le carbone 3 (C3). Ce groupe constitue la tête polaire et donc la partie hydrophile du cholestérol. La fonction -OH du cholestérol peut être estérifiée par un acide gras qui rend la molécule totalement insoluble dans l'eau.
Localisation
Le cholestérol est présent sous forme de stérides (cholestérol estérifié) dans la plupart des tissus des vertébrés, et en particulier le foie, le cerveau et la moelle épinière.
Rôle
C'est un composant majeur des membranes cellulaires animales qui contribue à leur stabilité et au maintien de leurs structures en s'intercalant entre les phospholipides (formant la bicouche de la membrane).
Il fluidifie la membrane car il empêche sa gélification en évitant la cristallisation des acides gras, et diminue la perméabilité membranaire aux molécules hydrosolubles.
Il a un rôle de « tampon thermique » : à 37 °C, il limite le mouvement des phospholipides, donc la fluidité membranaire diminue ; à des températures plus basses, il empêche l'entassement des phospholipides.
Dans la membrane, il permet la formation de radeaux lipidiques, zone essentielle à l'ancrage de protéines fonctionnelles.
Dans les neurones, il contribue à la libération des neurotransmetteurs lors de leur exocytose et donc la propagation de l'influx nerveux.
Le métabolisme du cholestérol est également précurseur de nombreuses molécules :
- les hormones stéroïdiennes : cortisol, cortisone et aldostérone ;
- les hormones stéroïdiennes sexuelles : progestérone, œstrogènes et testostérone ;
- le cholécalciférol (vitamine D3) ;
- l'hème A ;
- les protéines prénylées ou farnésylées ;
- l'ubiquinone ou coenzyme Q10 ;
- le dolichol ;
- le facteur nucléaire NF kappa B ;
- la protéine tau ;
- les sels biliaires.
Métabolisme
Synthèse
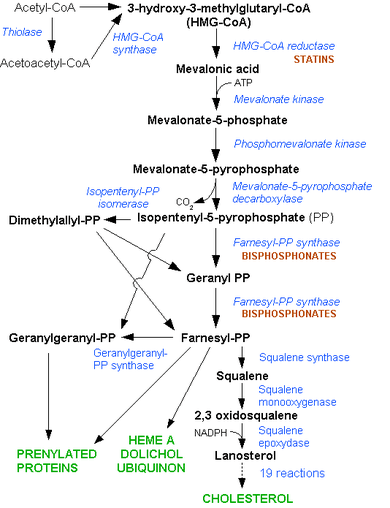
La synthèse du cholestérol se fait dans le cytoplasme des cellules du foie et de l'intestin principalement. La synthèse débute par la condensation de trois molécules d'acétate (=3×2 atomes de carbone) en hydroxy-méthyl-glutarate (ou HMG = 1×6 carbones). L'hydroxy-méthyl-glutarate est ensuite réduit en mévalonate (=1×6 carbones) par une enzyme, l'hydroxyméthylglutaryl-CoA réductase (ou HMGCoA réductase), en présence de coenzyme A. Cette étape est régulée par les statines. Le mévalonate est ensuite décarboxylé en isoprénoïdes à cinq carbones (l'isopentényl pyrophosphate et le diméthylallyl pyrophosphate). La condensation de six molécules d'isoprénoïdes aboutit au squalène (6×5=30 carbones). Enfin, le squalène subit l'action de la squalène cyclase qui crée les cycles du cholestérol à partir des insaturations présentes dans le squalène.
Régulation
Une partie du cholestérol peut ne pas être absorbée lors de la digestion. Par exemple, une bactérie intestinale présente chez l'homme transforme le cholestérol en coprostanol, stable et non absorbable et donc éliminée dans les selles[24]. Plus on mange de cholestérol et moins notre corps en absorbe, en pourcentage. C'est un premier niveau de régulation. Exemples : si vous mangez 400 mg de cholestérol, votre organisme l'absorbe à 40 %. Si vous en mangez 800 mg, votre organisme l'absorbe à 20 %.
Concernant le cholestérol absorbé ou produit par l'organisme, il existe trois niveaux de régulation du cholestérol, le but étant de diminuer le taux de cholestérol de la cellule quand il est en excès :
- le cholestérol libre dans la cellule inhibe la production de ses propres récepteurs membranaires LDLR. Pour ce faire, il inhibe la transcription du gène qui code les LDLR. Par conséquent, le flux entrant de cholestérol dans la cellule est diminué ;
- le cholestérol libre inhibe la HMG-coA réductase, ce qui empêche la poursuite de la réaction de synthèse du cholestérol ;
- enfin, le cholestérol libre stimule l’acyl transférase (ACAT), enzyme catalysant son estérification en stéride. Ceci favorise le stockage du cholestérol libre.
La synthèse de mévalonate, deuxième étape de la synthèse du cholestérol, est très régulée par le métabolisme. L'activité de l'HMG-CoA réductase, enzyme catalysant cette synthèse, est diminuée lorsque l'apport alimentaire en cholestérol est élevé ou par des médicaments de la famille des statines. Dans le cas d'une alimentation équilibrée, la proportion de cholestérol d'origine endogène est estimée entre 50 %[25] et 80 %[26] selon les auteurs (environ 700 mg/j), le reste du cholestérol étant d'origine alimentaire (entre 50 % et 20 % donc). L'augmentation des apports en cholestérol d'origine alimentaire inhibe la synthèse du cholestérol d'origine endogène.
À partir des éléments précédents, on peut dire que le cholestérol alimentaire influe très peu sur la cholestérolémie[27].
Dégradation
Le cholestérol est dégradé dans le foie en acides biliaires (dont l'acide chénodésoxycholique) par la 7-α-hydroxylase. La colestyramine[28], un médicament utilisé pour traiter l'hypercholestérolémie, diminue l'absorption intestinale des acides biliaires, et par conséquent leur concentration dans les cellules hépatiques. Ceci entraîne une activation de la 7-α-hydroxylase favorisant la dégradation du cholestérol.
Transport du cholestérol dans le sang : LDL, VLDL et HDL
En tant que composé hydrophobe, le cholestérol n'est pas soluble dans le sang. C'est pourquoi il est assimilé à une graisse, alors que c'est un stérol. Son transport est assuré par différents types de lipoprotéines :
- Les lipoprotéines de basse densité (ou LDL : low-density lipoprotein) transportent le cholestérol (ainsi que des triglycérides et des vitamines liposolubles) des lieux de sécrétion vers les cellules de l'organisme. Ces cellules expriment des récepteurs à leur surface pour indiquer leur besoin en cholestérol (ou autres substances).
Hypothèse du « mauvais » cholestérol : des taux importants de LDL conduiraient au dépôt de cholestérol sur les parois des artères (les récepteurs à LDL du foie et des tissus sont en effet très sensibles : au moindre changement biochimique d'une LDL, du fait d'une oxydation, d'une glycation ou d'une dégradation liée à la fumée de cigarette ou à d'autres facteurs comme le sucre, les lipoprotéines transportant le cholestérol ne seraient plus reconnues, et seraient donc phagocytées, puis formeraient un dépôt) sous forme de plaque d'athérome, ce qui pourrait accroître le risque de maladies cardiovasculaires et leur vaut le nom de « mauvais » cholestérol.
Cette hypothèse est une simplification de la réalité complexe des lipoprotéines. Ces différents transporteurs ne sont ni « bons », ni « mauvais » et s'échangent entre eux du cholestérol, ce qui fait qu'il n'y a pas de frontière réelle entre ces particules dans la réalité. Seul l'esprit humain essaie de classer ce qui forme un continuum de transporteurs de tailles diverses en des ensembles figés. Il existe d'ailleurs, selon la classification, différentes lipoprotéines de basse densité (LDL1, 2 et 3) et les plus dangereuses, selon le modèle actuel, sont les plus petites et les plus denses (LDL3). Ce seraient les plus athérogènes[29],[30].
- Les lipoprotéines de haute densité (ou HDL : high-density lipoprotein) déchargent les artères et les tissus extrahépatiques du cholestérol oxydé, et le ramènent vers le foie où il est dégradé ; on parle alors de « bon » cholestérol, même si cela désigne un cholestérol usé et qui va être recyclé.
- Les chylomicrons, ces lipoprotéines assurent le transport des lipides (cholestérol inclus) de l'intestin vers les autres tissus.
- Les lipoprotéines de très basse densité (ou VLDL : very low-density lipoprotein).
La classification tient également compte des apolipoprotéines : Apo A en relation avec les HDL, Apo B en relation avec les LDL. Apo E. On dose également la lipoprotéine(a) pour évaluer le risque vasculaire. Son rôle athérogène découle de sa capacité à se lier fortement à la paroi artérielle. L'augmentation du risque d'athérosclérose est proportionnelle à la concentration circulante de lipoprotéine(a)[31].
Teneur en cholestérol dans l'alimentation
| Aliment | Teneur en cholestérol (mg/100 g) |
Aliment | Teneur en cholestérol (mg/100 g) |
|---|---|---|---|
| cervelle de veau | 2 200 | ris de veau | 225 |
| jaune d'œuf | 300 | crème | 124 |
| rognons de mouton ou de veau | 380 | poulet | 90 à 100 |
| rognons de porc | 365 | fromage | 50 à 100 |
| foie de porc | 340 | veau | 84 |
| foie de veau | 314 | merlan | 77 |
| foie de bœuf | 265 | bœuf | 67 |
| beurre | 260 | poisson | 60 à 70 |
Ces teneurs en cholestérol alimentaire sont à compléter et relativiser par[32] :
- les teneurs en gras trans (pouvant favoriser l'oxydation des VLDL) ;
- les teneurs en acides gras mono-insaturés et poly-insaturés ;
- les teneurs en acides gras saturés ;
- l'apport alimentaire d'antioxydants (par exemple vitamine E, vitamine C, β-carotène) pouvant limiter l'accumulation des LDL dans la paroi artérielle.
Le tableau montre que toutes les viandes, mêmes maigres (abats, poulet, etc.) sont sources de cholestérol, en particulier les abats[33].
Le cholestérol présent dans les VLDL et LDL provient en effet des tissus (où il est excédentaire) qui l'ont synthétisé, et nullement des chylomicrons (structure de transport des lipides provenant de l'intestin). Limiter les apports alimentaires de cholestérol, ou son absorption au niveau de l'intestin (au travers de l'absorption intensive de phytostérols, par exemple), pour un individu ne souffrant pas d'hypercholestérolémie familiale, n'a par conséquent que peu d'effet prévention-santé.
Anomalies du dosage sanguin chez l'être humain
Le dosage du cholestérol sanguin se fait de manière traditionnelle chez un patient à jeun mais son taux global ainsi que sa fraction HDL ne sont pas modifiés de manière importante par le jeûne[34]. En médecine préventive, la Fédération française de cardiologie recommande de surveiller son taux de cholestérol à partir de dix-huit ans et tous les cinq ans[35].
Hypercholestérolémie et athérosclérose
Dès le début du XXe siècle, les travaux d’Anitschkow et Chalatow avaient permis de mettre en évidence un rôle du cholestérol dans l’athérosclérose expérimentale chez le lapin, en le nourrissant de graisses animales[36]. Aujourd'hui, plusieurs études proposent que l’athérosclérose soit une maladie inflammatoire[37],[38] et qu'un marqueur de cette maladie soit probablement le cholestérol associé aux LDL après oxydation[39]. Parallèlement, plusieurs études ont mis en évidence un lien entre hypercholestérolémie et présence d’une réaction inflammatoire dans le tissu vasculaire[40],[41].
Le taux de cholestérol est inclus dans plusieurs méthodes de calcul de ce risque (« échelle de risque »)[42] mais pas dans toutes[43].
Le troisième argument en faisant un facteur de risque cardio-vasculaire est l'existence d'essais cliniques de prévention primaire et secondaire chez les sujets hypercholestérolémiques qui ont démontré qu’il était possible de réduire la fréquence des cardiopathies ischémiques en diminuant le cholestérol associé au LDL à l’aide de statines[44]. Cependant d'autres études qui ont diminué le taux de cholestérol n'ont cependant pas montré de réduction de la mortalité globale (étude Helsinki avec le gemfibrozil, étude indépendante ALLHAT, étude LRC-CPPT, étude AFCAPS/TexCAPS) ou ont augmenté la mortalité globale (étude OMS-WHO avec le clofibrate, étude ILLUMINATE avec le torcetrapib[45]). De plus, les statines sont efficaces, quel que soit le niveau de cholestérol initial, et le niveau du cholestérol n'entre pas dans les critères d'entrée de la plupart des études[19]. Les statines jouant manifestement sur d'autres facteurs, se pose la question de savoir si le bénéfice est uniquement dû à la baisse du cholestérol. Quelques chercheurs, comme Michel de Lorgeril[46] et Uffe Ravnskov[47], ont ainsi remis en question « l'hypothèse lipidique » d'Ancel Keys, selon laquelle l'excès de consommation de graisses saturées et le cholestérol seraient les principaux responsables des maladies cardio-vasculaires[48].
D'autres facteurs influencent cependant le risque vasculaire, et jouent sur le taux de cholestérol. Ils rendent les données difficiles à interpréter : ainsi, l'activité physique régulière, l'alcool (à faible dose) et les œstrogènes pourraient contribuer à augmenter le taux de HDL, limitant le taux de cholestérol sanguin et améliorant la protection vasculaire[réf. nécessaire].
Inversement, le tabac, l'obésité et l'inactivité diminuent le taux de HDL sanguin, augmentant le risque cardio-vasculaire[réf. nécessaire].
Du point de vue diététique, les éléments a priori importants de ce point de vue, sur lesquels le régime peut jouer, sont les lipides et les glucides.
- Dans les années 1950-1960, l'Américain Ancel Keys développe « l'hypothèse lipidique » faisant un lien statistique (de corrélation) entre le taux de cholestérol sanguin et le risque de maladie cardio-vasculaire[49],[50].
- Une diminution des graisses animales est souhaitable : parmi les régimes testés, le régime méditerranéen diminue le risque cardiaque sans modifier le taux de cholestérol , mais ces régimes jouent aussi sur d'autres facteurs (obésité, diabète, etc.)[51].
- Le régime Low-carb est contre-productif : les régimes uniquement pauvres en glucides (dits « low-carb[52] ») semblent inefficaces à moyen et long terme, et dangereux ; ils augmentent le cholestérol et le risque cardiovasculaire selon une étude suédoise[53] longue (vingt-cinq ans), lancée en 1986 au nord de la Suède dans une région au risque cardiovasculaire élevé (comté de Västerbotten).
Cette étude est associée à un programme anti-obésité qui a permis dans un premier temps de diminuer les apports lipidiques moyens. Mais cette diminution a été suivie d'une stagnation (1992 à 2002) et d'une réaugmentation (jusqu'à un dépassement du niveau de 1986)[53]. Par contre la consommation de sucres a fortement baissé (après une hausse observée jusqu'en 1992), ce qui traduit selon les auteurs une appropriation par la population des régimes très médiatisés « low-carb » qui promeuvent de manger autant de lipides et protéines qu'on le souhaite mais en réduisant drastiquement l'apport en sucres. Ces régimes « low-carb » ont en fait augmenté la cholestérolémie moyenne (qui avait chuté au début du programme quand la consommation de lipides diminuait)[53]. Le poids moyen des habitants a en outre continué à augmenter de 1986 à 2012, malgré une perte effective mais provisoire de poids en début de régime[53]. Et la consommation de vin qui a (dans le panel suivi) augmenté de 1986 à 2010 (au détriment des alcools forts, avec augmentation parallèle mais moindre de la consommation de bière, uniquement chez les hommes) ne semble avoir eu aucun effet (chez l'homme ou la femme) sur la cholestérolémie ni sur le risque cardiovasculaire[54]. En outre la cholestérolémie augmente depuis les années 2000 malgré une augmentation significative de médicaments visant à la faire diminuer[55].
Défaut de synthèse
- La chondrodystrophie calcifiante congénitale est une maladie génétique provoquant une altération du fonctionnement de la 3-β-hydroxystéroïde-δ(8), δ(7)-isomérase, une enzyme impliquée dans la synthèse du cholestérol.
- Le syndrome de Smith-Lemli-Opitz est une maladie génétique provoquant un déficit en 7-déhydrocholestérol réductase, l'enzyme responsable de la dernière étape de la synthèse du cholestérol.
Prix Nobel concernant le cholestérol
L'étude de cette molécule a à trois reprises été récompensée par des prix Nobel :
- en 1964, Konrad Bloch et Feodor Lynen reçurent le prix Nobel de médecine pour « leur découverte concernant le mécanisme de régulation des métabolismes du cholestérol et des acides gras »[56] ;
- en 1965, Robert Burns Woodward reçut le prix Nobel de chimie pour « ses exceptionnelles réalisations dans l'art de la synthèse organique »[57]. Il a été le premier à réaliser la synthèse chimique du cholestérol et de la cortisone en 1951 ;
- en 1985, Michael S. Brown et Joseph L. Goldstein reçurent le prix Nobel de médecine pour « leur découverte portant sur la régulation du métabolisme du cholestérol »[58].
Notes et références
- Masse molaire calculée d’après « Atomic weights of the elements 2007 », sur www.chem.qmul.ac.uk.
- Entrée de « Cholesterol » dans la base de données de produits chimiques GESTIS de la IFA (organisme allemand responsable de la sécurité et de la santé au travail) (allemand, anglais), accès le 24 juin 2009 (JavaScript nécessaire).
- « Cholesterol », sur www.reciprocalnet.org (consulté le 12 décembre 2009).
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, « Evaluations Globales de la Cancérogénicité pour l'Homme, Groupe 3 : Inclassables quant à leur cancérogénicité pour l'Homme », sur http://monographs.iarc.fr, CIRC, (consulté le 22 août 2009).
- Karl Feltgen, Le Cholestérol : 1758-1913. Essai historique sur l'intérêt qu'il a suscité en médecine depuis sa découverte au milieu du XVIIIe jusqu'à l'aube du XXe siècle, Thèse Médecine, Rouen, 1993, .
- (en) Tibblin G, Wilhelmsen L, Werkö L, « Risk factors for myocardial infarction and death due to ischemic heart disease and other causes », American Journal of Cardiology, vol. 35, no 4, , p. 514-522.
- Année qui voit une nouvelle technique d'analyse disponible, capable de différentier le LDL et HDL.
- (en) Peter W. F. Wilson, Ralph B. D’Agostino, Daniel Levy, Albert M. Belanger, Halit Silbershatz and William B. Kannel, « Prediction of Coronary Heart Disease Using Risk Factor Categories », Circulation, vol. 9, , p. 1837–1847 (DOI 10.1161/01.CIR.97.18.1837).
- (en) Ancel Keys, « Coronary heart disease in seven countries », Circulation, no 41, , p. 1-211.
- Ce lien statistique ne signifie pas causalité.
- (en) Stefan Hofvendahl, « Coronary Heart Disease in Seven Countries. Edited by Ancel Keys, Ph.D. », Acta Medica Scandinavica, vol. 190, nos 1-6, , p. 464-464 (DOI 10.1111/j.0954-6820.1971.tb07460.x).
- (en) Uffe Ravnskov, The Cholesterol Myths: Exposing the Fallacy that Saturated Fat and Cholesterol Cause Heart Disease, Newtrends Publishing, , p. 320.
- (en) Michael Oliver, « The clofibrate saga: a retrospective commentary », British Journal of Clinical Pharmacology, vol. 74, no 6, , p. 907-910 (DOI 10.1111/j.1365-2125.2012.04282.x).
- (en) « WHO cooperative trial on primary prevention of ischaemic heart disease with clofibrate to lower serum cholesterol : final mortality follow-up. Report of the Committee of Principal Investigators », Lancet, vol. 2, no 8403, , p. 600–604.
- (en) Cristin E. Kearns, Laura A. Schmidt, Stanton A. Glantz, « Sugar Industry and Coronary Heart Disease Research. A Historical Analysis of Internal Industry Documents », JAMA Internal Medicine, (DOI 10.1001/jamainternmed.2016.5394).
- Renaud Roussel, Du beurre, s'il vous plaît, Grancher, , p. 47.
- (en) Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4 444 patients with coronary heart disease : the Scandinavian Simvastatin Survival Study (4S), Lancet, 1994;344:1383-1389.
- (en) Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, McKillop JH, Packard CJ, « Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group », The New England Journal of Medicine, no 333, , p. 1301-1307.
- (en) Norbert Donner-Banzhoff et Andreas Sönnichsen, « Strategies for prescribing statins », BMJ, vol. 336, , p. 288-289.
- (en) M. de Lorgeril, P. Salen, J.-L. Martin, I. Monjaud, J. Delaye et N. Mamelle, « Mediterranean Diet, Traditional Risk Factors, and the Rate of Cardiovascular Complications After Myocardial Infarction : Final Report of the Lyon Diet Heart Study », Circulation, vol. 99, no 6, , p. 779-785 (DOI 10.1161/01.CIR.99.6.779).
- (en) Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III), JAMA, 2001;285:2486-97.
- Cholestérol : le grand bluff, Documentaire réalisé par Anne Georget, France, 2016, Arte.
- Dominique Dupagne, émission La Tête au carré sur France Inter, 22 février 2013.
- Bactérie trouvée par des chercheurs de Jouy-en-Josas. Gérard P., Lepercq P., Leclerc M., Gavini F., Raibaud P., Juste C., Bacteroides sp. Strain D8, the First Cholesterol-Reducing Bacterium Isolated from Human Feces, Applied and Environmental Microbiology, 73:5742-5749, 2007.
- Biochimie de Harper, 25e éd., De Boeck édition, 2002, chap. 28.
- Biochimie et biologie moléculaire, Omniscience édition, 2006, chap. 19.
- EUFIC : Conseil européen de l'information sur l'alimentation.
- Banque de Données Automatisée sur les Médicaments : « Colestyramine », 13 octobre 2000.
- Bernard Chanu, « Rôle athérogène des lipoprotéines riches en triglycérides », Médecine thérapeutique/Endocrinologie, vol. 4, , p. 199-207 (lire en ligne).
- http://archive-ouverte.unige.ch/downloader/vital/pdf/tmp/3bfulh529jlitbkg99aabf8t20/out.pdf.
- http://www.admp.fr/aide-decision-medicale/424/dosage-de-la-lp-a-1ere-determination-biochimie-.htm.
- www.objectif-equilibre-sante.info, « Diététique préventive cardio-vasculaire », 14 novembre 2006.
- http://www.uvp5.univ-paris5.fr/campus-nutrition/cycle1/Poly/0800fra.asp.
- Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report, Circulation, 2002;106:3143–3421 (p. 3170).
- Le cholestérol sur le site de la Fédération française de cardiologie.
- (de) (en) Anitschkow N., Chalatow S., « On experimental cholesterin steatosis and its significance in the origin of some pathological processes », Centralblatt fur Allgemeine Pathologie und Pathologische Anatomie, vol. 24:1–9, 1913 (trad. dans Arteriosclerosis, vol. 3:178–182, 1983.
- (en) Davies M.J., « Stability and instability : Two faces of coronary atherosclerosis », Circulation, vol. 94:2013-2020, 1996.
- (en) Ross R., « Atherosclerosis-an inflammatory disease », N. Engl. J. Med., vol. 340:115-126, 1999.
- (en) Steinberg D., Parthasarathy S., Carew T.E., Khoo J.C., Witztum J.L., « Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity », N. Engl. J. Med., vol. 320:915-924, 1989.
- (en) Liao E., Andalibi A., Qiao J.H., Allayee H., Fogelman A.M., Lusis A.J., « Genetic evidence for a common pathway mediating oxidative stress, inflammatory gene induction, and aortic fatty streak formation in mice », J. Clin. Invest., vol. 94:877-884, 1994.
- (en) Young S.G., « Parthasarathy S.- Why are low-density lipoproteins atherogenic ? », West. J. Med., vol. 160:153-164, 1994.
- Conroy RM, Pyörälä K, Fitzgerald AP et al., the SCORE project group, Estimation of ten-year risk of fatal cardiovascular disease in Europe : the SCORE project, Eur. Heart J., 2003;24:987-1003.
- Gaziano TA, Young CR, Fitzmaurice G, Atwood S, Gaziano JM, Laboratory-based versus non-laboratory-based method for assessment of cardiovascular disease risk : the NHANES I Follow-up Study cohort, Lancet, 2008;371:923-931.
- (en) Steinberg D., Gotto A. M. Jr., « Preventing coronary artery disease by lowering cholesterol levels: fifty years from bench to bedside », JAMA, vol. 282:2043-2050, 1999.
- http://www.theheart.org/article/758713.do.
- Michel de Lorgeril, 2008, Cholestérol, mensonges et propagande, Thierry Souccar Éditions (ISBN 978-2-916878-17-1).
- The Cholesterol Myths: Exposing the Fallacy that Saturated Fat and Cholesterol Cause Heart Disease / Ravnskov, Uffe (ISBN 0-9670897-0-0).
- (de) (fr) Gebbers J.L. (trad. Legoy M.), « Le cholestérol n'a rien à voir avec le développement de l'athérosclérose », Ars Medici, vol. 9(98): S564-569.
- (en) Keys A., Taylor H.L., Blackburn H., Brozek J., Anderson J.T., Simonson E., « Coronary heart disease among Minnesota business and professional men followed 15 years », Circulation, vol. 28:381-95, 1963.
- (en) Keys A., « Seven countries : a multivariate analysis of death and coronary heart disease », Harvard University Press, Londres, 1980.
- Kastorini CM, Milionis HJ, Esposito K et al., The Effect of Mediterranean Diet on Metabolic Syndrome and its Components : A Meta-Analysis of 50 Studies and 534,906 Individuals, J. Am. Coll. Cardiol., 2011 57: 1299-1313.
- Régimes dits « Low carbohydrate high-fat » ou « LCHF » pour les anglophones.
- Romain Loury, Cholestérol : les régimes « low-carb » démasqués, Journal de l'environnement, 20 juin 2012, article relatif à une étude : Associations among 25-year trends in diet, cholesterol and BMI from 140,000 observations in men and women in Northern Sweden ; publiée dans Nutrition Journal 2012, DOI:10.1186/1475-2891-11-40 et notamment suivie par l'université d'Umea (ISSN 1475-2891), URL, 27 p..
- Voir figure 6 associée à la première présentation de l'étude déjà citée.
- Voir figure 7 associée à la première présentation de l'étude déjà citée.
- (en) nobelprize.org, « The Nobel Prize in Physiology or Medicine 1964 ».
- (en) nobelprize.org, « The Nobel Prize in Chemistry 1965 »
- (en) nobelprize.org, « The Nobel Prize in Physiology or Medicine 1985 ».
Voir aussi
Articles connexes
- Hypercholestérolémie
- Lipide
- Acide gras
- Sténose
- Radeau lipidique
Bibliographie
- Drs Philippe Even, Bernard Debré, La Vérité sur le Cholestérol, Le Cherche midi, 2013
- Dr Michel de Lorgeril. Cholestérol, Mensonges et Propagande, Thierry Souccar Éditions, 2013
- Nouvelle Société Française d'Athérosclérose, « Athérosclérose:physiopathologie »
- (en) Center for Disease Control and Prevention, « Ancel Keys, Ph.D. », Morbidity and Mortality Weekly Report, vol. 48 (30):651, 1999
- Dr Feltgen, Poulletier de la salle et la découverte du cholestérol [PDF], Groupe d'histoire des Hôpitaux de Rouen



