Cardiomyopathie dilatée
Une cardiomyopathie dilatée (CMD) est une forme de cardiomyopathie (littéralement, maladie du muscle cardiaque) dans laquelle les cavités cardiaques (principalement les ventricules) sont dilatées (à différents degrés), diminuant de façon significative la capacité du muscle cardiaque à assurer sa fonction de "pompe", conduisant ainsi à l'insuffisance cardiaque et couplé à un risque de mort subite, quel que soit le stade de la maladie.
| Spécialité | Cardiologie |
|---|
| CIM-10 | I42.0 |
|---|---|
| CIM-9 | 425.4 |
| OMIM | 212110 |
| DiseasesDB | 3066 |
| MedlinePlus | 000168 |
| eMedicine | 152696 et 895187 |
| eMedicine | med/289 emerg/80 ped/2502 |
| MeSH | D002311 |
| GeneReviews | et NBK1119 Dilated Cardiomyopathy Overview |
![]()
Une cause ischémique (séquelles d'un infarctus du myocarde ou conséquence d'atteintes sévères des artères coronaires) est habituellement exclue de cette entité.
Cette dernière est la forme la plus courante de cardiomyopathie.
Épidémiologie
La cardiomyopathie dilatée touche plus fréquemment les hommes que les femmes, entre 20 et 60 ans, avec un pic de fréquence entre 20 et 40. Mais elle peut également être rencontrée chez les enfants. Sa prévalence est de un cas sur 2 500 personnes avec des formes familiales dans presque un cas sur deux[1]. La cause des cardiomyopathies dilatées dites "secondaires" la plus fréquemment rencontrée en France est l'alcoolisme chronique.
Physiopathologie
L'atteinte du muscle cardiaque diminue sa force contractile et entraîne, à terme, une diminution du débit cardiaque avec ses conséquences sur l'organisme (tableau d'insuffisance cardiaque).
Il existe un certain nombre de mécanismes compensateurs à cet état qui aboutit à un « remodelage » des cavités cardiaques dont le ventricule gauche. Celui-ci augmente son volume maximal (en fin de diastole, ou télédiastolique, quand il est le plus rempli) et son volume télésystolique (en fin de systole ; volume minimum quand le ventricule gauche a éjecté le sang dans l'aorte). La différence entre volume télédiastolique et télésystolique correspond au volume d'éjection systolique (VES), qui conditionne le débit cardiaque. On l'apprécie par la fraction d'éjection (rapport du volume d'éjection sur le volume télédiastolique).
Cette augmentation des volumes sanguins va compenser, dans un premier temps, selon une loi dite « loi de Starling » la diminution de la contractilité myocardique permettant ainsi une conservation du débit cardiaque. Ce remodelage va s'accompagner d'une modification de ses parois, habituellement un amincissement, d'une modification de la forme du ventricule qui devient plus sphérique et moins allongé. Ce processus va aboutir à un déclin progressif de la fraction d'éjection. Ce concept de remodelage cardiaque a initialement été développé pour décrire les modifications intervenant dans les jours et les mois suivant un infarctus du myocarde. Il a été étendu aux cardiomyopathies non ischémiques, telles que la cardiomyopathie dilatée idiopathique ou la myocardite chronique, suggérant des mécanismes communs dans la progression du dysfonctionnement cardiaque.
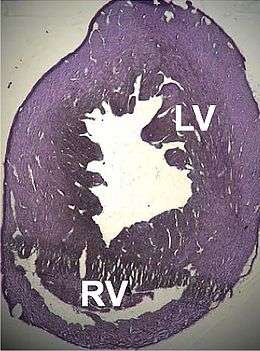
Anatomiquement, le cœur est plus gros, ses cavités (principalement le ventricule gauche) sont dilatées, les anneaux valvulaires sont eux aussi dilatés.
Causes
Dans la majorité des cas, la cause de la cardiomyopathie dilatée n'est pas connue : on parle alors de forme idiopathique ou primitive.
Les lésions myocardiaques peuvent être d'origine génétique (formes familiales), toxique (drogues (cocaïne), alcool, chimiothérapie anticancéreuse, imipraminiques[2]), métabolique (hyperthyroïdie), infectieux (infection à virus : Coxsackie B, Rickettiose, mononucléose… parasitoses : maladie de Chagas…), auto-immune (lupus érythémateux disséminé, sclérodermie, périartérite noueuse…), sarcoïdose, liée à une cardiomyopathie extrinsèque (ischémique, hypertensive, rythmique…).
Le syndrome de tako-tsubo, véritable sidération myocardique survenant après un stress émotionnel[3] en est une forme particulière.
La cardiomyopathie du peripartum survient typiquement un mois avant l'accouchement et jusqu'à cinq mois après[4]. Son mécanisme n'est pas clair.
Environ 20 à 40 % des formes sont dites familiales, liées à des mutations de gènes codant diverses protéines présentes dans les cellules musculaires myocardiques[5],[6]. La maladie est génétiquement hétérogène, mais le plus souvent, il s'agit d'une transmission autosomique dominante (comme dans le Syndrome d'Alström). Des formes autosomiques récessives, à transmission liée à l'X (dominante ou récessive) ou à transmission mitochondriale ont également été décrites.
Cardiomyopathies génétiques
Les formes familiales constitueraient entre 30 et 50 % des cas[7]. Les mutations responsables concernent les gènes intervenant dans la synthèse du cytosquelette ou de la fibre musculaire sarcomère). Plus de 60 gènes responsables ont été identifiés, les plus couramment mis en cause étant TTN, MYH7, TNNT3, TNNI3, SDP, LMNA et SCN5A[8]. La recherche d'une cause génétique n'est pas faite de manière systématique si le cas est isolé.
| Gène | Protéine | Pourcentage | OMIM | Pathologies en rapport |
|---|---|---|---|---|
| ACTC1 | Actine | < 1 % | 102540 | Cardiomyopathie hypertrophique |
| DES | Desmine | < 1 % | 1125660 | Myopathie avec surcharge en desmine |
| LMNA | Lamine | 7 % - 8 % | 150330 | Dystrophie musculaire d'Emery-Dreifuss Maladie de Charcot-Marie-Tooth type 2 Progéria |
| SGCD | Delta-sarcoglycane | ? | 601411 | élément |
| MYH7 | Myosine 7 | 5 % - 8 % | 160760 | Myopathie distale de type Laing |
| TNNT2 | Troponine T | 2 % - 4 % | 191045 | élément |
| TPM1 | Tropomyosine alpha-1 | ? | 191010 | élément |
| TTN | Titine | ? | 188440 | Myopathie tibiale de Udd |
| VCL | Vinculine | ? | 193065 | |
| MYBPC3 | élément | élément | élément | élément |
| PLN | élément | élément | élément | élément |
| LDB3 | élément | élément | élément | élément |
| ACTN2 | élément | élément | élément | élément |
| CSRP3 | élément | élément | élément | élément |
| MYH6 | élément | élément | élément | élément |
| ABCC9 | élément | élément | élément | élément |
| TNNC1 | élément | élément | élément | élément |
| TCAP | élément | élément | élément | élément |
| SCN5A | élément | élément | élément | élément |
| EYA4 | élément | élément | élément | élément |
| TMPO | élément | élément | élément | élément |
| PSEN1 | élément | élément | élément | élément |
| PSEN2 | élément | élément | élément | élément |
| FCMD | Fukutine | ? | 607440 | Dystrophie musculaire de Fukuyama |
| Gène | Protéine | OMIM | Pourcentage | Pathologies en rapport |
|---|---|---|---|---|
| DMD | Dystrophine | 300377 | Myopathie de Duchenne de Boulogne | |
| TAZ | Tafazine | < 1 % | 300394 | Syndrome de Barth Fibroélastose de type 2 |
Certaines mutations présentent un tableau plus particulier : troubles du rythme ou de la conduction chez les patients porteurs d'une atteinte dès gènes LMNA ou SCN5A.
Diagnostic
Signes fonctionnels
Pour certains sujets, la cardiomyopathie dilatée ne va pas entraîner de signes fonctionnels majeurs (voire pas de signes du tout) et donc ne pas ou peu retentir sur la qualité de vie, ni sur la durée de vie. La maladie peut donc être découverte de façon fortuite, lors de la réalisation de certains examens (électrocardiogramme, radiographie pulmonaire).
Cependant, il va pouvoir exister un certain nombre de symptômes. Le symptôme majeur est la dyspnée d'effort (essoufflement lors de la réalisation d'efforts plus ou moins importants). Peuvent s'y associer une asthénie, des palpitations, de vagues douleurs thoraciques insensibles à la trinitrine (Des douleurs typiques d'angine de poitrine sont inhabituelles et doivent, dans ce cas, suggérer la coexistence d'une cardiopathie ischémique), des malaises et syncopes, des manifestations thrombo-emboliques et parfois un risque de mort subite.
Des signes d'insuffisance cardiaque droite et gauche vont pouvoir se développer progressivement. À noter qu'une dilatation du ventricule gauche peut être présente depuis des mois (parfois même des années) avant qu'un sujet ne devienne symptomatique.
Examen clinique
L'examen clinique va pouvoir retrouver une tachycardie, un bruit de galop, un souffle cardiaque d'insuffisance mitrale ou tricuspide fonctionnel (qui n'est pas dû à une lésion organique des valves cardiaques), des signes d'insuffisance cardiaque.
À un stade avancé de la maladie, la pression artérielle est basse.
Examens complémentaires
Une radiographie pulmonaire va pouvoir mettre en évidence, dans les formes déjà évoluées, une augmentation de taille de la silhouette cardiaque (cardiomégalie) importante (index cardio-thoracique supérieur à 0,60 dans 30 % des cas). La dilatation cardiaque apparaît globale (bien qu'intéressant principalement le ventricule gauche). Elle peut également découvrir des anomalies liées au bas débit cardiaque (œdème pulmonaire), épanchement pleural...
L'électrocardiogramme ne montre pas d'anomalie spécifique. Il sert à éliminer un infarctus du myocarde et à dépister des complications comme des troubles du rythme cardiaque (fibrillation auriculaire, arythmies ventriculaires...) ou de la conduction. Il peut cependant être normal.
L'échographie cardiaque est l'examen essentiel au diagnostic. Il montre une dilatation cardiaque globale avec une épaisseur de paroi normale ou amincie, des diamètres télédiastoliques augmentés, une diminution globale de la contraction cardiaque qui peut être quantifier par la mesure de la fraction d'éjection, plus ou moins diminuée (inférieure à 60-75 %). Cet examen permet en outre de suivre l'évolution de la maladie.
Une coronarographie (examen radiologique d'opacification des artère coronaires) peut être réalisée afin d'éliminer une cardiopathie ischémique. L'injection du produit de contraste dans le ventricule gauche permet également de calculer la fraction d'éjection. Cet examen peut être couplée avec la mesure des pressions intracardiaques et pulmonaires : Il existe une élévation de la pression télédiastolique du ventricule gauche et une augmentation des pressions capillaires et artérielles pulmonaires. Le débit cardiaque et le volume d'éjection systolique sont altérés (sauf au début de la maladie).
Des explorations isotopiques (gamma-scintigraphie au Technétium, scintigraphie myocardique au Thallium, IRM cardiaque) peuvent apporter des renseignements sur les volumes des cavités cardiaques, la contractilité cardiaque et la fraction d'éjection.
Une biopsie myocardique est exceptionnellement réalisée. Elle permet le diagnostic des cardiopathies de surcharge, causes rares de cardiomyopathies, telles que la sarcoïdose ou l'hémochromatose. Dans ces cas, un traitement spécifique peut être proposé. Dans d'autre cas, elle peut retrouver le virus en cause d'une myocardite dont le cardiomyopathie constitue la séquelle.
Il n'existe pas à ce jour de test génétique prédictif pour cette maladie.
Prise en charge
Il est souvent prescrit le traitement standard de l'insuffisance cardiaque : régime pauvre en sel, inhibiteurs de l'enzyme de conversion de l'angiotensine, diurétiques, bêta-bloquants, digitaliques ou amiodarone en cas d'arythmie.
Les anticoagulants sont parfois également utilisés en cas de manifestations thrombo-emboliques, surtout si une fibrillation auriculaire est associée.
Certains patients bénéficient de l'implantation d'un stimulateur cardiaque multi site, voire de défibrillateurs implantables en cas de risque d'arythmie ventriculaire.
Pour les patients à un stade avancé et qui deviennent réfractaires au traitement médical, une transplantation cardiaque est à discuter.
Évolution et complications
L'évolution est variable et se fait classiquement vers l'insuffisance cardiaque (incapacité du cœur à assurer un débit cardiaque efficace pour des efforts de plus en plus restreints), que le sujet tente de compenser en réduisant son activité physique (réduction délétère).
Elle est très souvent émaillée de poussées aiguës d'insuffisance cardiaque (œdème aigu du poumon).
Ces poussées, pratiquement constantes dans l'évolution peuvent être liées à "des facteurs déclenchant" : existence de troubles du rythme (passage en tachy-arytmie complète par fibrillation auriculaire), surcharge sodée (non-respect du régime sans sel, excès accidentels des repas de fins d'année...), maladies intercurrentes (grippe, broncho-pneumopathie...), anémie... Ces poussées réagissent habituellement aux traitements habituels.
La répétition des poussées sans facteur déclenchant est un élément de mauvais pronostic, faisant redouter l'insuffisance cardiaque "terminale" (dyspnée permanente au repos empêchant de dormir en décubitus (orthopnée) - position allongée - et obligeant à vivre pratiquement sans bouger, en position demi-assise).
Il existe certains cas de réversibilité en fonction de la cause de la cardiomyopathie dilatée (alcool, cocaïne, médicaments, hyperthyroïdie, syndrome de tako-tsubo…).
Les principales complications sont les manifestations thrombo-emboliques (embolie systémique ou pulmonaire), les troubles du rythme cardiaque (fibrillation auriculaire, extrasystoles ventriculaires, tachycardie ventriculaire), avec un risque non négligeable de mort subite.
Chez les animaux
Cette section ne cite pas suffisamment ses sources (juillet 2013). Pour l'améliorer, ajoutez des références vérifiables [comment faire ?] ou le modèle {{Référence nécessaire}} sur les passages nécessitant une source. |
La cardiomyopathie dilatée est une maladie existant également chez certaines races de chiens[9] : Boxer, Dobermann, Dogue allemand, Saint-bernard.
Cette pathologie touche également certaines races de chats[réf. souhaitée].
Elle est également rencontrée de temps en temps chez les rongeurs[réf. souhaitée] tels que les rats à la suite des nombreux problèmes respiratoires qu'ils peuvent rencontrer.
Notes et références
- (en) Taylor MR, Carniel E, Mestroni L. « Cardiomyopathy, familial dilated » Orphanet J Rare Dis. 2006;1:27
- Revue Prescrire. « Cardiomyopathie dilatée liée aux antidépresseurs imipraminiques ? » Rev Prescrire 2003;23.833.
- Muriel Gevrey, « Le tako-tsubo : un diagnostic différentiel à évoquer devant un syndrome coronaire aigu à coronaire saine », sur http://www.theheart.org, (consulté le 21 novembre 2009)
- (en) Hilfiker-Kleiner D, Sliwa K, Drexler H. « Peripartum cardiomyopathy: recent insights in its pathophysiology » Trends Cardiovasc Med. 2008;18:173-9.
- (en) Schonberger J. Seidman C.E. « Many roads leads to a broken heart : the genetics of dilated cardiomyopathy » Am J Hum Gen. 2001;69:249-60.
- (en) Ross J. Jr. « Dilated cardiomyopathy : concepts derived from gene deficient and transgenic animal models » Circ J. 2002;66:219-224.
- (en) Jefferies JL, Towbin JA. « Dilated cardiomyopathy » Lancet 2010;375: 752-62.
- Japp AG, Gulati A, Cook SA, Cowie MR, Prasad SK, The diagnosis and evaluation of dilated cardiomyopathy, J Am Coll Cardiol, 2016;67:2996-3010
- (en) Oyama M.A. Chittu S. « Genomic expression patterns of cardiac tissues from dogs with dilated cardiomyopathy » Am J Vet Res. 2005;66:1140-55.
- (en) Cet article est partiellement issu d’une traduction de l’article en anglais : "Dilated cardiomyopathy".
Voir aussi
Articles connexes
Liens externes
- Pr Pony. Les myocardiopathies (Site médical Réseau pédagogique de la Faculté de Médecine de Rennes) - Site visité le 18/03/2007.
- (en)Site "Cardiomyopathy Association" - Cardiomyopathie dilatée (en) - Site visité le 18/03/2007.
