Hémophilie
L'hémophilie est une anomalie constitutionnelle de la coagulation sanguine en rapport avec un déficit d’un des facteurs de la coagulation. Cependant, dans un tiers des cas, l'hémophilie est engendrée par une mutation de novo. Ces défauts sont dus à une déficience d'un des facteurs suivants : XI, IX ou VIII, ou à la présence d'anticoagulants contre l'un de ces facteurs.
| Spécialité | Hématologie |
|---|
| CIM-10 | D66-D68 |
|---|---|
| CIM-9 | 286 |
| OMIM | 306700 306900 264900 |
| DiseasesDB | 5555 5561 29376 |
| MedlinePlus | 000537 |
| eMedicine | 779322 |
| eMedicine | med/3528 |
| MeSH | D025861 |
![]()
Les manifestations cliniques de la maladie sont proportionnelles au déficit du facteur de la coagulation. Les manifestations cliniques correspondent aux hémorragies qui peuvent atteindre chaque organe, en particulier les articulations (hémarthroses) et les muscles (hématomes). La maladie peut être sévère avec manifestations dès la première année de vie, ou légère avec très peu de manifestations.
Il existe plusieurs types d’hémophilie en rapport avec le facteur de coagulation déficitaire : l’hémophilie A (« hémophilie classique ») correspond à une mutation du gène du facteur VIII, l’hémophilie B (ou Christmas disease) correspond à une mutation du gène du facteur IX, et l’hémophilie C (ou « maladie de Rosenthal ») correspond à une mutation du gène du facteur XI.
Histoire
Cette maladie est connue depuis des millénaires. Les Hébreux de l'Antiquité la connaissaient déjà et ne pratiquaient jamais la circoncision des garçons dont la mère était issue d'une famille ayant perdu un enfant par hémorragie lors d'une circoncision. Elle fut aussi appelée « maladie royale », étant donné que la reine Victoria du Royaume-Uni a transmis l’hémophilie aux familles royales d’Espagne, d’Allemagne et de Russie. On notera que sans cette maladie, Grigori Raspoutine n'aurait jamais été aussi célèbre qu'il l'a été. Ce dernier aurait réussi à soulager le tsarevitch Alexis (fils de Nicolas II) de ce mal que les médecins de l'époque connaissaient peu[1]. En effet la médecine de l'époque ignorait les propriétés de l'aspirine qui était donnée au jeune malade pour soigner ses douleurs. Ce médicament est un antiagrégant plaquettaire, facteur donc aggravant de l'hémophilie. Le simple fait de balayer de la table et de jeter les « remèdes » donnés au malade – dont l'aspirine – ne pouvait qu'améliorer son état[2]. Raspoutine réussit ainsi à se faire passer pour une personne ayant des pouvoirs mystiques.
L'épidémie du sida a été particulièrement meurtrière pour les hémophiles. Ceux-ci, ayant besoin de transfusions régulières, ont été nombreux à contracter le virus. En France, cela a donné lieu à un grand scandale politique : l'affaire du sang contaminé.
Types
- Hémophilie A : Mutation du gène F8 du locus q28 du chromosome X codant le facteur VIII de coagulation. Cette mutation consiste souvent en une inversion de l'intron 22 soit des délétions et insertions. L'incidence de cette maladie est de 1 sur 5 000 naissances de garçons avec une prévalence de 1 sur 10 000 dans les pays à haut équipement sanitaire. Elle peut être confondue avec la Maladie de Willebrand car celle-ci présente aussi une diminution du facteur VIII.
- Hémophilie B : Mutation du gène F9 du locus q27 du chromosome X codant le facteur IX de coagulation. Il existe plus de 2 100 mutations pouvant porter sur ce gène. L'incidence de cette maladie est de 1 sur 20 000 naissances de garçons avec une prévalence de 1 sur 25 000 dans les pays à haut équipement sanitaire. Elle peut également être appelée « maladie de Christmas ».
- Hémophilie C : L'hémophilie C est un déficit en facteur XI. Elle atteint environ une personne sur 100 000[3]. Il s'agit d'une forme légère d'hémophilie non liée au sexe et donc touchant les hommes et les femmes. Le plus souvent elle ne nécessite pas de traitement. Les femmes sont affectées plus sévèrement du fait des menstruations.
Dans de très rares cas, des hémophilies plus spécifiques peuvent être observées, par exemple l'hémophilie B Leyden, qui se résorbe progressivement à partir de la puberté pour finir sur un taux de 30 à 70 %.
Diagnostic
Signes cliniques
Le diagnostic de l'hémophilie repose sur les examens courants pratiqués dans l’exploration de l’hémostase. Le temps de céphaline activé est augmenté ; le taux des facteurs de coagulation VIII, IX ou XI est abaissé ; le temps de saignement est le plus souvent normal ; le taux de prothrombine et la numération plaquettaire sont normaux.
Le sang d'une personne atteinte d'hémophilie ne coagule pas normalement. Les saignements ne sont pas plus abondants, ni plus rapides que la normale, mais durent plus longtemps. Les blessures superficielles sont généralement bénignes et ne provoquent pas de saignement plus intense que la normale. D'autres saignements peuvent entraîner des conséquences plus graves, en particulier s'ils concernent les articulations, surtout les genoux, les chevilles et les coudes, ainsi que les tissus mous et les muscles. La gravité de l'hémophilie est en fonction du pourcentage d’activité du facteur de coagulation déficitaire.
Examens complémentaires
- Allongement isolé du TCA: c'est-à-dire: Plaquettes = Normales; Test de Quick = Normal; Temps de Thrombine = Normal
- Test de correction positive
- Dosages spécifique des facteurs de coagulation plasmatique: Dosage du Facteur VIII (FVIII) puis du Facteur IX (FIX). L'activité normale d'un humain est située entre 50 et 150 % de taux de référence - le témoin normal étant à 100 % par définition. L'hémophilie est qualifiée de « mineure » à « sévère », selon le taux du facteur de coagulation déficient, mais également de la potentialité à faire des hémarthroses spontanées (sans aucun mouvement, un « épanchement » de sang survient au niveau de l'articulation, attaquant le cartilage). Dans des cas d'hémophilie mineure, entre 5 et 30 % d’activité du facteur de coagulation. Les symptômes incluent : aucune apparition de trouble de saignement, aucun saignement sauf dans le cas d’une blessure, risque de saignement prolongé après une intervention chirurgicale ou une lésion grave ; et hémorragies rares. Dans des cas d'hémophilie modérée, entre 1 et 5 % d’activité du facteur de coagulation. Les symptômes incluent : risque de saignement prolongé après une intervention chirurgicale ; lésion grave ou intervention dentaire ; présence d'épisodes de saignement environ une fois par mois ; et saignements rarement voire jamais sans raison évidente. Dans des cas d'hémophilie sévère, moins de 1 % d’activité du facteur de coagulation. Les symptômes incluent : hémorragies fréquentes au niveau des muscles ou des articulations (principalement les genoux, les coudes et les chevilles) ; présence d'épisodes de saignement une ou deux fois par semaine ; et des saignements sans raison évidente.
- Rechercher la présence éventuelle d'un inhibiteur du F VIII ou du F IX, titrer cet inhibiteur si nécessaire
Autres syndromes hémophiliques
Les patients peuvent avoir d'autres déficits héréditaires en facteur de coagulation :
- Déficit en Facteur VII. Transmission autosomique récessive, prévalence : 1/500 000[4].
- Déficit en Facteur II. Transmission autosomique récessive.
- Déficit en Facteur V. Transmission autosomique récessive, prévalence : 1/1 000 000.
- Déficit en Facteur X. Transmission autosomique récessive, prévalence : 1/1 000 000[5].
- Afibrinogénémie (déficit en fibrinogène ou facteur I). Transmission autosomique récessive; prévalence : 5/10 000 000.
- Déficit combiné en Facteur V et Facteur VIII. Transmission autosomique récessive ; très rare, situé sur le chromosome 18.
- Déficit en Facteur XIII. Transmission autosomique récessive, extrêmement rare[6].
- Déficit congénital en vitamine K[7].
- Les déficits en Facteur XII, en prékallicréine ou en Kininogène de haut poids moléculaire ne provoquent pas de saignements mais un allongement du TCA[8].
Ils peuvent également avoir des déficits acquis en facteur de coagulation :
- Insuffisance hépatique
- CIVD
- Déficit en vitamine K, anti-vitamine K
- Anticorps anti-facteurs de coagulation[9],[10].
De plus, bien que n'étant pas considérés comme hémophilies, les troubles de l'hémostase primaire et les excès de fibrinolyse mènent aussi à un risque hémorragique.
Épidémiologie
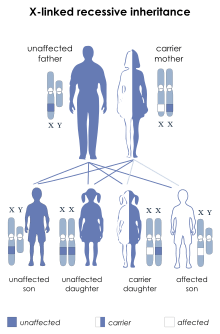
Les hémophilies A et B sont récessives et liées au chromosome X mais un tiers des hémophilies correspondent à une mutation de novo[réf. nécessaire]. On observe donc qu'un homme qui porte le Xh est toujours atteint par la maladie alors que la femme n'est que porteuse (mais pourra la transmettre à ses descendants). Il faut en tenir compte pour le conseil génétique.
Des cas rares de femmes hémophiles (Xh/Xh) ont été répertoriés. Il pourrait s'agir de femmes issues de l'union d'un père hémophile (Xh Y) et d'une mère porteuse (X Xh), mais la probabilité d'une telle ascendance demeure faible. Des cas ont également été répertoriés où la femme ne présente qu'un seul X touché (Xh X) et manifeste cependant une hémophilie sévère ; cela est dû à un trouble grave de l'inactivation du chromosome X sain[11],[12]. Par ailleurs des femmes porteuses peuvent aussi présenter des symptômes plus ou moins graves : on les appelle « porteuses à taux faible ».
Traitements
Les traitements actuels ne guérissent pas de l'hémophilie mais ils consistent en l'administration par voie intraveineuse de Facteur VIII ou de Facteur IX permettant d'obtenir une activité coagulante suffisante pour arrêter, voire prévenir, l’hémorragie. Les traitements actuels sont soit issus du plasma sanguin (produits plasmatiques, ex. : Factane (facteur VIII), Betafact (facteur IX), soit des produits synthétiques issus de biotechnologie (produits recombinants), ayant partiellement (ex. : Kogenate, Helixate, Refacto) ou totalement (ex. : Advate, Benefix) éliminé les traces de dérivés sanguins de leurs procédés de fabrication et dans le produit final. L'élimination des dérivés sanguins permet de supprimer les risques de transmission de certaines maladies du donneur (SIDA, maladies virales). Ces molécules ont une durée de vie courte (moins de 48 h pour la plus longue[13]), obligeant à en renouveler l'administration en cas de problème hémorragique persistant et ne pouvant, en aucun cas, constituer un traitement de fond. De plus, la transfusion itérative de facteurs de coagulation peut entraîner, dans certains cas, la formation d'anticorps contre ces derniers, les rendant inefficaces[14] et nécessitant une prise en charge plus complexe.
Le laboratoire français du fractionnement et des biotechnologies conçoit des traitements contre l'hémophilie.
Pistes de recherche
l'emcizumab est un anticorps monoclonal ayant une double spécificité pour le facteur IX activé et le facteur X, permettant le rapprochement de ces deux molécules et jouant ainsi le rôle d'un facteur VIII. En injection sous-cutanée hebdomadaire, il diminue très sensiblement le risque d'hémorragie[15].
Un ARN interférent contre le gène de l'antithrombine (SERPINC1) a été développé et en cours de test dans le traitement de l'hémophilie[16].
L'injection d'un virus au génotype modifié pour contenir le gène du facteur VIII[17] ou le facteur IX[18] permet une normalisation du taux de ces facteur ans la plupart des cas et la quasi-disparition du risque hémorragique, avec un recul de plus d'un an.
Notes et références
- (en) Kendrick, J.M. « Russia's imperial blood: was Rasputin not the healer of legend? » Am J Hematol. 2004;77(1):92-102
- (en) Jeffreys Diarmuid, Aspirin. The Remarkable Story of a Wonder Drug, Bloomsbury Publishing,
- « Déficit en facteur XI (Hémophilie C) », lire en ligne sur hemophilia.ca
- http://www.ima.org.il/imaj/ar08june-22.pdf
- (en) « Factor X deficiency: clinical manifestation of 102 subjects from Europe and Latin America with mutations in the factor 10 gene. » (consulté le 11 août 2014).
- (en) « Factor XIII deficiency in children--clinical presentation and outcome. » (consulté le 11 août 2014).
- (en) « Inherited vitamin K deficiency: case report and review of literature. » (consulté le 11 août 2014).
- (en) « Hemophilia A. Synonyms: Classic Hemophilia, Factor VIII Deficiency » (consulté le 11 août 2014).
- (en) « A monoclonal IgG4 (lambda) with factor V inhibitory activity. » (consulté le 11 août 2014).
- (en) « Gross hematuria due to acquired haemophilia in hereditary hemorrhagic telangiectasia. » (consulté le 11 août 2014)
- « Comment les femelles « font taire » de manière réversible un de leurs deux chromosomes sexuels ? », sur cnrs.fr (consulté le 13 août 2014).
- « L’hémophilie » [PDF] (consulté le 13 août 2014).
- Tiede A, Half-life extended factor VIII for the treatment of hemophilia A, J Thromb Haemost, 2015;13:Suppl 1:S176-S179
- Witmer C, Young G, Factor VIII inhibitors in hemophilia A: rationale and latest evidence, Ther Adv Hematol, 2013;4:59-72
- Oldenburg J, Mahlangu JN, Kim B et al. Emicizumab prophylaxis in hemophilia A with inhibitors, N Engl J Med, 2017;377:809-818
- Pasi KJ, Rangarajan S, Georgiev P et al. Targeting of antithrombin in hemophilia A or B with RNAi therapy, N Engl J Med, 2017;377:819-828
- Rangarajan S, Walsh L, Lester W et al. AAV5–factor VIII gene transfer in severe hemophilia A, N Engl J Med, 2017;377:2519-2530
- George LA, Sullivan SK, Giermasz A et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant, N Engl J Med, 2017;377:2215-2227
Article connexe
- L'hémophilie dans la descendance de Victoria
Liens externes
- AMA-Contacts (Bulletin de l'Association des Médecins Anciens Étudiants de l'Université de Louvain, Belgique) Numéro 30, juin 2003
- (en) GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993-2005 Hémophilie A sur genetests.org
- (en) GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993-2005 Hémophilie B sur genetests.org
- Site d'information généraliste destiné aux patients hémophiles et à leur entourage
- L'hémophilie sur Sante-AZ

