Déficit en mévalonate kinase
Le déficit en mévalonate kinase (ou MKD pour mevalonate kinase deficiency)[1], encore nommé acidurie mévalonique (mevalonic aciduria pour les anglophones[2]) est une maladie rare autosomale récessive (environ 200 cas répertoriés dans le monde) dite MVK.
| Spécialité | Hématologie, neurologie, immunologie, génétique médicale et endocrinologie |
|---|
| OMIM | 251170 |
|---|---|
| DiseasesDB | 29843 |
| MeSH | D054078 |
![]()
La maladie
C'est une maladie métabolique rare, caractérisée par une impossibilité de normalement biosynthétiser le cholestérol et les isoprénoïdes[3], en raison d'une déficience en une enzyme, la Mévalonate kinase, codée par le gène MVK.
 cycle du mévalonate
cycle du mévalonate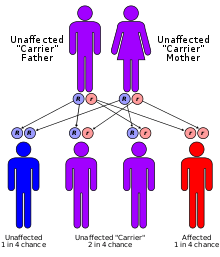 Transmission de type autosomale récessive
Transmission de type autosomale récessive Acide mévalonique
Acide mévalonique
Histoire
Ce désordre métabolique et le syndrome dont il est responsable ont été pour la première fois médicalement décrit en 1985[4].
Présentation clinique
La maladie débute généralement dans l'enfance, avec des épisodes de 3 à 6 jours de fièvre, qui seront récurrents.
La fréquence de retour des crises varie selon les individus.
Cette fréquence est généralement plus élevée dans l'enfance (25 fois par an en moyenne) et diminue à l'âge adulte[1].
- Une forme moins sévère est dite hyperimmunoglobulinemia D syndrome (HIDS)
- une forme plus dure est dite Acidurie mévalonique (mevalonic aciduria ou MVA).
Symptômes
Ce sont typiquement :
- des ganglions gonflés (lymphadénopathie) ;
- des douleurs abdominales ;
- une hépatosplénomégalie
- des douleurs articulaires ;
- des diarrhées ;
- des rashs sur la peau (urticaire) ;
- des maux de tête ;
- teint pâle (en raison d'une anémie) ;
et parfois apparaissent des aphtes buccaux (ou vaginaux chez la femme).
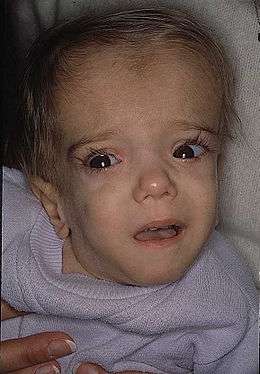
Quelques patients développent des malformations congénitales, des troubles mentaux ou un retard de développement mental marqué et/ou psychomoteur (ataxie, hypotonie, des problèmes d'yeux et des crises d'épilepsie[1]).
Rarement, une amyloïdose entraîne une déficience rénale qui peut conduire à la mort.
Facteurs de déclenchement de crise
Les crises sont plus ou moins récurrentes, mais une vaccination, un acte chirurgical, une blessure ou un stress peuvent aussi les déclencher[1].
Diagnostic
Les analyses d'urine révèlent une accumulation d'acide mévalonique (en raison du déficit en enzyme mévalonate kinase[5] (ATP:mevalonate 5-phosphotransferase; EC 2.7.1.36).
Les taux sanguin d'immunoglobuline D (IgD) et d'immunoglobuline A (IgA) sont souvent anormalement élevés (pour des raisons mal comprises, et sans relations apparentes avec les symptômes)[1].
Pour les patients atteint de la forme bénigne (HID), entre les crises la vie est normale, et l'espérance de vie ne semble pas diminuée[1].
Ceux qui sont affectés de la forme dure (MVA), ont une qualité de vie très dégradée et une morphologie anormale (tête plus petite et allongée)[1]. Hormis pour les cas les plus graves, l'espérance de vie semble normale[1].
Voir aussi
Articles connexes
Liens externes
- (fr) ANGIS Database, Mevalonate kinase deficiency
- Cette section est vide, insuffisamment détaillée ou incomplète. Votre aide est la bienvenue ! Comment faire ?
Bibliographie
- Cette section est vide, insuffisamment détaillée ou incomplète. Votre aide est la bienvenue ! Comment faire ?
Notes et références
- NIH (Gouvernement des États-Unis, Genetic Conditions ; Mevalonate kinase deficiency
- (en) 251170
- (en) Mancini J, Philip N, Chabrol B, Divry P, Rolland MO, Pinsard N, « Mevalonic aciduria in 3 siblings: a new recognizable metabolic encephalopathy », Pediatr. Neurol., vol. 9, no 3, , p. 243–246 (PMID 8352861, DOI 10.1016/0887-8994(93)90095-T)
- (en) Berger R, Smit GP, Schierbeek H, Bijsterveld K, le Coultre R, « Mevalonic aciduria: an inborn error of cholesterol biosynthesis? », Clin. Chim. Acta, vol. 152, nos 1-2, , p. 219–222 (PMID 4053401, DOI 10.1016/0009-8981(85)90195-0)
- (en) Bretón Martínez JR, Cánovas Martínez A, Casaña Pérez S, Escribá Alepuz J, Giménez Vázquez F, « Mevalonic aciduria: report of two cases », J. Inherit. Metab. Dis., vol. 30, no 5, , p. 829 (PMID 17578678, DOI 10.1007/s10545-007-0618-7)
