Formule de Clapeyron
En chimie physique, et plus particulièrement en thermodynamique, la formule de Clapeyron, également appelée relation de Clapeyron[1] ou équation de Clapeyron, est une relation exprimant l'évolution de la pression de changement d'état d'un corps pur en fonction de la température, voir la figure 1. Elle porte le nom d'Émile Clapeyron qui l'établit en 1834[2].
Ne doit pas être confondu avec Formule de Clausius-Clapeyron ou Relations de Clapeyron.
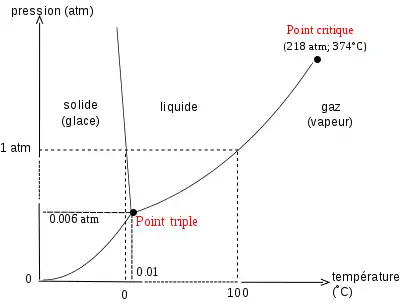
Note : ce diagramme est celui de l'eau, qui présente la particularité d'avoir une branche d'équilibre solide-liquide de pente négative ; pour la plupart des espèces chimiques cette branche a une pente positive.
Cette formule n'est valable que pour une transition de phase d'ordre un selon la classification d'Ehrenfest des changements d'état, c'est-à-dire, selon la classification actuelle, pour un changement d'état impliquant une enthalpie de changement d'état. Si tel n'est pas le cas, la transition est d'ordre supérieur et il faut se rapporter aux formules d'Ehrenfest. La formule de Clapeyron est donc principalement valable pour les transitions liquide-gaz, solide-liquide et solide-gaz.
La formule de Clausius-Clapeyron (ou relation de Clausius-Clapeyron, équation de Clausius-Clapeyron) est une forme simplifiée de la formule de Clapeyron permettant son intégration dans le cas d'un équilibre liquide-vapeur. Elle fut établie par Clapeyron dès 1834 et retrouvée par Rudolf Clausius en 1850[3].
La formule de Clapeyron permet, entre autres, la détermination expérimentale de l'enthalpie de changement d'état.
La formule de Clapeyron peut être étendue aux mélanges.
Énoncé et démonstration
Diagramme de Phase

Les principales transitions de phase impliquant une enthalpie de changement d'état sont les transitions liquide-gaz, solide-liquide et solide-gaz, voir la figure 2. Les changements d'état se faisant à pression constante pour une température donnée, on peut reporter dans un diagramme P-T les domaines d'existence des diverses phases, voir la figure 1. La formule de Clapeyron représente la pente des courbes délimitant ces domaines dans ce diagramme.
Les trois courbes de transition se rejoignent au point triple qui est le seul point où coexistent les trois états physiques liquide, solide et gaz du corps pur. La branche de vaporisation se termine au point critique, point au-delà duquel le liquide et le gaz ne peuvent plus être différenciés.
Énoncé
À température donnée, un changement d'état d'un corps pur d'une phase notée à une autre notée s'effectue à pression constante , ce que l'on représente sur un diagramme de phase. Pour une transition de phase d'ordre un selon la classification d'Ehrenfest des changements d'état, la pression de changement d'état varie en fonction de la température selon la formule de Clapeyron[4] :




| Formule de Clapeyron : |

ou :

avec :
- la température de changement d'état (en kelvins, K) ;
- la pression de changement d'état à la température (en pascals, Pa) ;
- l'enthalpie de changement d'état de la phase à la phase à la température (en joules par mole, J/mol) ;
- , avec , l'entropie de changement d'état de la phase à la phase à la température (en joules par mole, J/mol) ;
- le volume de changement d'état, c'est-à-dire la différence des volumes molaires du corps pur respectivement dans les phases et à la température et sous la pression (en mètres cubes par mole, m3/mol).




On peut également écrire cette formule selon[5] :

avec :
- la différence des facteurs de compressibilité du corps pur respectivement dans les phases et , et , soit (ces grandeurs sont adimensionnelles) ;
- la constante universelle des gaz parfaits.





- Note 1 - unité de l'enthalpie de changement d'état
- L'enthalpie de changement d'état peut être également exprimée par unité de masse (par ex. en J/kg), auquel cas il faut considérer des volumes massiques (en m3/kg) au dénominateur.
- Note 2 - transition inverse
- Pour une transition de la phase à la phase on considère et . Par conséquent :



- Les transitions de l'état 1 vers l'état 2 et de l'état 2 vers l'état 1 ayant pour point commun le point triple, ceci implique qu'il ne peut y avoir d'hystérésis dans les changements d'état d'un corps pur : pour une température donnée , autrement dit, que la transition se fasse dans un sens ou dans l'autre la pression de changement d'état d'un corps pur est la même.

- Note 3 - transitions de phase d'ordre supérieur
- Cette formule n'est valable que dans le cas d'une transition de phase du premier ordre selon la classification d'Ehrenfest des changements d'état. Pour les transitions de phase du deuxième ordre, voir les formules d'Ehrenfest.
Classification d'Ehrenfest, transition d'ordre un
La classification d'Ehrenfest des transitions de phase repose sur le principe suivant :
« Une transition de phase est d'ordre n si la fonction enthalpie libre et ses dérivées jusqu'à l'ordre n-1 sont continues, tandis qu'une de ses dérivées d'ordre n au moins est discontinue. »
Pour une transition d'ordre 1, la dérivée d'ordre 0 de l'enthalpie libre est continue lors du changement d'état. Autrement dit, l'enthalpie libre est invariante lors du changement d'état à pression et température constantes. Ainsi, dans les conditions d'équilibre (mêmes pression et température), l'enthalpie libre d'une quantité de corps pur dans l'état est égale à l'enthalpie libre de cette même quantité de corps pur dans l'état :



Étant donné l'identité de l'enthalpie libre molaire d'un corps pur et de son potentiel chimique, ceci implique que :

En revanche, pour cette même transition d'ordre 1, les dérivées partielles d'ordre 1 de l'enthalpie libre :


ne sont pas continues :


avec :
- et les volumes de la quantité du corps pur respectivement dans les phases et , à la température et sous la pression ;
- et les entropies de la quantité du corps pur respectivement dans les phases et , à la température et sous la pression .




On a donc, en termes de volume molaire et d'entropie molaire :


Dans une transition de phase d'ordre 1 il y a donc une enthalpie de changement d'état :

Dans une transition de phase d'ordre un le corps pur change de volume (par exemple dans la vaporisation d'un liquide le volume résultant de gaz est supérieur au volume initial de liquide), contrairement à une transition de phase d'ordre deux (par exemple la transition conducteur-supraconducteur) où le volume du corps pur ne change pas. De plus, une transition de phase d'ordre un induit un changement d'entropie du corps pur, et donc implique une enthalpie de changement d'état (la vaporisation d'un liquide nécessite l'apport de chaleur), contrairement à une transition d'ordre deux où il n'y a pas d'entropie de changement d'état. Dans une transition d'ordre un le corps pur passe par une série d'états intermédiaires où les deux phases coexistent (dans une vaporisation le liquide et la vapeur coexistent depuis l'état de liquide seul jusqu'à l'état de vapeur seule tant que la chaleur totale nécessaire à son évaporation complète n'a pas été apportée au liquide) ; au contraire les deux phases ne coexistent pas dans une transition d'ordre deux, celle-ci étant immédiate.
Démonstration
Considérons le changement d'état d'un corps pur défini par l'équation suivante, mettant en jeu les phases et à pression et température constantes :


À l'équilibre des phases, les potentiels chimiques du corps pur dans les deux phases sont égaux :

Si l'on modifie la température initiale de l'équilibre pour une autre température , tout en restant à l'équilibre des deux phases, alors la pression d'équilibre passe de à et les potentiels chimiques passent respectivement de à et de à . Les potentiels chimiques des deux phases sont toujours égaux lorsque le système atteint son nouvel équilibre. On peut écrire pour le nouvel équilibre :







d'où l'égalité des variations des deux potentiels chimiques :

Selon la relation de Gibbs-Duhem appliquée à moles de corps pur, la variation du potentiel chimique vaut :


avec :
- le volume molaire du corps pur à et ;
- l'entropie molaire du corps pur à et .



Il s'ensuit, en déclinant l'expression pour chacune des deux phases et , et en considérant l'égalité des variations des potentiels chimiques :


On considère ici une transition de phase de premier ordre selon la classification d'Ehrenfest, soit :
ce qui permet d'écrire :

En introduisant l'enthalpie de changement d'état , l'entropie de changement d'état du corps pur vaut :

En notant le volume de changement d'état :
on obtient finalement la formule de Clapeyron :
Relations de Clapeyron intégrées
On intègre entre deux états d'équilibre impliquant les deux mêmes phases : un état de référence et un état .


L'enthalpie de changement d'état ne dépend que de la température. On peut par approximation considérer l'enthalpie de changement d'état comme constante sur des intervalles de température de l'ordre de quelques dizaines voire centaines de degrés.
Les volumes molaires dépendent de la température et de la pression. Dans le cas des phases condensées (liquide et solide) cette dépendance est faible et peut être considérée comme négligeable pour des intervalles de température peu importants. Les volumes molaires des phases condensées sont en outre négligeables devant celui d'une phase gaz, à condition toutefois d'être suffisamment éloigné du point critique pour un liquide.
Transition liquide-gaz, formule de Clausius-Clapeyron
Considérons la vaporisation d'un liquide :

L'enthalpie de changement d'état mise en jeu est l'enthalpie de vaporisation . L'écart des volumes molaires vaut .


Si l'on est suffisamment loin du point critique du corps pur, le volume molaire du liquide est négligeable devant celui du gaz[6] :


D'autre part, à des pressions de l'ordre de la pression atmosphérique le gaz peut être considéré comme parfait, aussi son volume molaire peut-il être calculé selon la loi des gaz parfaits :

avec la constante universelle des gaz parfaits. On obtient la formule de Clausius-Clapeyron[7] :
| Formule de Clausius-Clapeyron : |

que l'on trouve également sous la forme :

Contrairement à la formule de Clapeyron qui est valable quelles que soient les conditions de pression et température, c'est-à-dire du point triple au point critique dans le diagramme de phase du corps pur, cette relation n'est donc valable que :
- si le volume molaire du liquide est négligeable devant celui du gaz, c'est-à-dire pour un équilibre loin du point critique,
- si le gaz se comporte comme un gaz parfait, c'est-à-dire pour un équilibre aux basses pressions.
On intègre (pour mémoire, une enthalpie de changement d'état ne dépend que de la température) :

La pression de vaporisation est le plus souvent appelée pression de vapeur saturante du corps pur, notée .


Si l'on considère l'enthalpie de vaporisation comme une constante, on obtient la formule de Rankine[8] :
| Formule de Rankine : |

ou, de façon plus générale :

En introduisant une troisième constante pour ajuster plus précisément la corrélation à des données expérimentales[9],[10], on obtient l'équation d'Antoine :


L'enthalpie de vaporisation est une grandeur positive, en conséquence la pression de vapeur saturante d'un corps augmente avec la température ; réciproquement, sa température de vaporisation (ébullition) augmente avec la pression. Par exemple l'eau bout à 100 °C sous la pression de 1 atm, mais au sommet du Mont Blanc, où la pression n'est que de 0,5 atm, l'eau bout à 85 °C. Au sommet de l'Everest, elle bout à 72 °C. Au contraire, au fond des océans la pression est telle que l'eau peut se maintenir liquide à plus de 300 °C près de sources chaudes[11].
Transition solide-gaz
Considérons la sublimation d'un solide :

L'enthalpie de changement d'état mise en jeu est l'enthalpie de sublimation . De même que pour l'équilibre liquide-gaz, le volume molaire du solide est négligeable devant celui du gaz, et aux basses pressions le gaz peut être considéré comme parfait. On obtient par conséquent, en intégrant la formule de Clapeyron en considérant l'enthalpie de sublimation constante, une formule similaire à la formule de Rankine obtenue précédemment :


L'enthalpie de sublimation étant supérieure à l'enthalpie de vaporisation, soit , aux alentours du point triple la courbe d'équilibre solide-gaz du corps pur dans un diagramme P-T a une pente supérieure à celle de la courbe d'équilibre liquide-gaz (voir figure 1).

Transition liquide-solide
Considérons la fusion d'un solide :

L'enthalpie de changement d'état mise en jeu est l'enthalpie de fusion . L'écart des volumes molaires vaut . Les deux phases condensées ont des volumes molaires ( pour le liquide et pour le solide) dépendant peu de la température et de la pression. On peut ainsi intégrer en considérant l'enthalpie de fusion et les volumes molaires comme constants :






et finalement :

La courbe associée à cette relation correspond à la branche d'équilibre liquide-solide sur le diagramme de phase. Cette branche est presque linéaire aux basses pressions et est pratiquement verticale à cause du faible écart des volumes molaires des phases condensées ( d'où et ).



L'enthalpie de fusion étant une grandeur positive et le volume molaire d'un liquide étant le plus souvent supérieur à celui d'un solide, pour la quasi-totalité des corps purs la pente de la branche liquide-solide du diagramme P-T est positive : . L'eau est une exception (ainsi que l'antimoine, l'argent, le bismuth, le gallium, le germanium, le plutonium et le sodium) car le volume molaire du solide (glace) est supérieur à celui du liquide (, par conséquent la glace flotte), ainsi , la pente de la branche d'équilibre solide-liquide de l'eau est négative : (voir la figure 1).




Applications
Détermination de l'enthalpie de vaporisation

La température et la pression d'équilibre associée d'un gaz au contact de son liquide peuvent être mesurées expérimentalement dans un autoclave. On peut ainsi déterminer l'évolution de la pression d'équilibre ou pression de vapeur saturante en fonction de la température. En considérant que l'enthalpie de vaporisation est constante sur l'intervalle de température expérimental, selon la formule de Rankine :


d'où :

Si l'on trace le graphe on obtient une droite de pente négative, voir figure 3 :


La détermination graphique de cette pente permet le calcul de l'enthalpie de vaporisation :

Cas particulier de la fusion de l'eau

L'eau est l'un des rares corps dont la masse volumique du solide est inférieure à celle du liquide (d'où le fait que la glace flotte). En conséquence, la température de fusion de l'eau diminue sous l'effet de la pression (pour la grande majorité des corps elle augmente avec la pression).
À pression atmosphérique la température de fusion de la glace est de = 0 °C (273,15 K). La formule de Clapeyron intégrée permet de calculer le changement de pression nécessaire pour faire baisser la température de fusion à = −1 °C (272,15 K) :


En utilisant les valeurs[12] :
- = 333,5 kJ/kg l'enthalpie de fusion massique de l'eau ;
- = 1,002 × 10−3 m3/kg le volume massique de l'eau liquide à 0 °C (soit une masse volumique de 998 kg/m3) ;
- = 1,090 5 × 10−3 m3/kg le volume massique de l'eau solide (glace) à 0 °C (soit une masse volumique de 917 kg/m3) ;



la pression relative à appliquer pour faire baisser la température de fusion de l'eau à −1 °C est de :
- = 13,82 MPa = 138,2 bar

soit une pression absolue de 139,2 bar.
Ceci est mis en évidence dans l'expérience de Tyndall (voir figure 5). Un fil lesté d'un poids est posé sur un pain de glace. Sous l'effet de la pression due au fil la température de fusion baisse et la glace fond localement, en conséquence le fil descend dans le bloc de glace. L'eau redevient solide après le passage du fil, lorsque la pression redevient normale. Le fil traverse ainsi le bloc de glace sans le rompre[13].
Cette particularité explique le déplacement des glaciers : le poids de la glace exerce sur la base du glacier une pression telle que l'eau y fond, permettant le glissement de la masse de glace par gravité sur un sol pentu. De même, en patinage sur glace l'augmentation de pression au sol (par rapport à la pression atmosphérique) due au poids du patineur fait baisser la température de fusion de la glace, celle-ci fond en conséquence et il se forme une mince couche d'eau liquide sous le patin, ce qui permet la glisse. Le poids du patineur étant transmis au sol par la surface de contact de la lame du patin, très faible, la pression résultante est très importante.
Météorologie
En météorologie, la formule de Clausius-Clapeyron est utilisée couramment dans les diagrammes thermodynamiques comme les téphigrammes, Skew-T et émagrammes pour le calcul des énergies de changement de phase de l'eau atmosphérique (notée ). Sur un diagramme pression-température (P-T), la ligne séparant les deux phases est la courbe de coexistence . Pour la pression de vapeur saturante de l'eau , la formule de Clausius-Clapeyron devient[14] :




où est la constante universelle des gaz parfaits.
Extension aux mélanges
Diagramme de phase des mélanges

Pour les mélanges on peut, à composition donnée, tracer les courbes de changement d'état dans un diagramme représentant la pression en fonction de la température. La figure 5 montre un ensemble de courbes d'équilibre liquide-vapeur du système méthane-éthane obtenues pour diverses compositions du mélange. Les courbes du méthane et de l'éthane purs y sont également représentées.
Les points critiques sont marqués par des cercles. Le lieu géométrique des points critiques des mélanges commence et finit aux points critiques des corps purs.
Une courbe pleine, pour une composition donnée, présente deux branches qui se rejoignent au point critique. La branche joignant le point critique par la gauche (basses températures) est la courbe d'ébullition du mélange ; celle joignant le point critique par la droite (hautes températures) est la courbe de rosée. À gauche de la courbe d'ébullition le mélange est liquide, à droite de la courbe de rosée il est gazeux. Au-delà du point critique il est supercritique.
Pour une composition donnée, entre la courbe d'ébullition et la courbe de rosée le mélange est biphasique. Dans ce domaine, à pression et température données, les compositions de la phase liquide et de la phase gaz en équilibre sont données respectivement par la courbe d'ébullition et la courbe de rosée passant par ce point du diagramme.
Comme pour un corps pur, on peut établir théoriquement l'expression des pentes de ces courbes d'ébullition et de rosée : il s'agit de l'extension de la formule de Clapeyron aux mélanges. Cette formule s'applique à tous les changements de phase entre solide, liquide et gaz.
Énoncé de la formule pour les mélanges
La formule de Clapeyron étendue aux mélanges[15],[16],[17], pour un mélange de constituants notés , pour une phase de composition constante en équilibre avec une phase de composition variable, s'écrit :


| Formule de Clapeyron étendue aux mélanges : |

ou :

avec :
- et les fractions molaires du corps respectivement dans les phases et ;
- l'enthalpie molaire de la phase ;
- et les enthalpies molaires partielles du corps respectivement dans les phases et ;
- le volume molaire de la phase ;
- et les volumes molaires partiels du corps respectivement dans les phases et .








- Note 1 - grandeurs molaires partielles de la phase
- Dans l'expression de , les grandeurs molaires partielles de la phase , et , sont pondérées par la composition de la phase , les fractions molaires . Néanmoins, elles sont des fonctions de la composition de la phase , les fractions molaires : et .
- Les grandeurs molaires de la phase , et , sont des fonctions de la composition de la phase , les fractions molaires : et .
- Les écarts et correspondent donc aux variations de propriétés d'un mélange de composition passant d'un état initial ayant les propriétés de l'état à un état final ayant les propriétés de l'état .











- Note 2 - formule du corps pur
- Dans le cas d'un corps pur les fractions molaires . Les grandeurs molaires partielles se confondent avec les grandeurs molaires : et . On retrouve la formule pour les corps purs.



- Note 3 - transition inverse
- Pour la transformation inverse, dans laquelle la phase à composition constante est en équilibre avec la phase de composition variable :

- avec :
- l'enthalpie molaire de la phase ;
- le volume molaire de la phase .


- À la température , on considère une phase et une phase de même composition :
- la phase est en équilibre avec une phase de composition à pression ,
- la phase est en équilibre avec une phase de composition à pression .


- On a :


- Ainsi la variation de la pression de changement d'état d'un mélange n'est-elle pas la même selon le sens de la transformation :

- En conséquence, pour un équilibre liquide-vapeur d'un mélange de composition donnée à température donnée, la pression de rosée (pression à laquelle apparait la première goutte de liquide dans le gaz) n'est pas identique à la pression de bulle (pression à laquelle apparait la première bulle de gaz dans le liquide). Les deux courbes se rejoignent cependant au point critique et si le système présente un azéotrope : dans les deux cas les deux phases ont la même composition .

Démonstration de la formule étendue
On suppose une phase en équilibre avec une phase . On étudie cet équilibre à composition de la phase constante. En modifiant la pression et la température, seule la composition de la phase est donc modifiée. Les deux phases sont des mélanges de composants repérés par l'indice . On note :
- et les fractions molaires du corps respectivement dans les phases et ;
- et les enthalpies molaires respectives des phases et ;
- et les enthalpies molaires partielles du corps respectivement dans les phases et ;
- et les entropies molaires respectives des phases et ;
- et les entropies molaires partielles du corps respectivement dans les phases et ;
- et les volumes molaires respectifs des phases et ;
- et les volumes molaires partiels du corps respectivement dans les phases et ;
- et les potentiels chimiques du corps respectivement dans les phases et .








À l'équilibre des phases on a les relations, pour chaque composant :
- (1)

Puisque l'on fait varier la pression, la température et la composition tout en restant à l'équilibre des phases, les potentiels chimiques varient, pour chaque composant, selon :
- (2)

La relation de Gibbs-Duhem donne pour la phase :

En substituant la relation (2) pour chaque composant on a :
- (3)

Pour la phase dont la composition est constante, on a pour chaque composant la relation :

En substituant cette relation pour chacun des constituants dans la relation (3) on obtient :
- (4)



Le théorème d'Euler implique que :

ainsi :
- (5)

Étant donné les relations entre grandeurs molaires partielles :


en considérant la relation (1) :


Par conséquent, la relation (5) devient :

Le théorème d'Euler implique que :

d'où :

En substituant cette relation dans la relation (4) et en considérant que la démonstration est faite à composition de la phase constante, on obtient la formule de Clapeyron étendue aux mélanges :
Application aux équilibres de mélanges idéaux
Si les deux phases en équilibre sont des mélanges idéaux, les enthalpies molaires partielles et les volumes molaires partiels se confondent respectivement avec les enthalpies molaires et les volumes molaires des corps purs aux mêmes pression, température et phases, soit :




En conséquence :

Les corps purs n'existent pas nécessairement dans les phases et dans les conditions de pression et de température du mélange. Néanmoins, pour les phases condensées (liquide et solide) il peut être considéré que les propriétés de ces phases sont indépendantes de la pression, elles sont généralement données en fonction de la température seule : elles peuvent en conséquence être utilisées à la température du mélange mais pour des pressions auxquelles les phases condensées n'existent pas. Pour un gaz, le gaz idéal est un gaz parfait qui peut exister à toute pression et toute température ; de plus l'enthalpie d'un gaz parfait ne dépend pas de la pression en vertu de la loi de Joule-Thomson. On note, pour tout corps :


La formule de Clapeyron étendue aux mélanges idéaux s'exprime selon :

avec :
- l'enthalpie de changement d'état du corps pur à la température ;
- l'écart des volumes molaires respectifs du corps pur dans les phases et à la température ; pour un gaz parfait .



Pour la pression de bulle d'un mélange liquide idéal, la phase est une phase gaz, la phase la phase liquide. L'enthalpie de changement d'état d'un corps pur est donc son enthalpie de vaporisation :


Au basses pressions, le volume molaire d'une phase condensée est négligeable devant celui d'un gaz en équilibre, aussi puisque la phase est un mélange de gaz parfaits :



De même, pour la pression de rosée d'un mélange gazeux idéal, la phase est une phase liquide, la phase la phase gaz. L'enthalpie de changement d'état d'un corps pur est donc son enthalpie de liquéfaction :


Pour les volumes molaires aux basses pressions, celui du liquide étant négligeable devant celui du gaz : .

On a par conséquent les formules de Clausius-Clapeyron pour les pressions de bulle et de rosée d'un mélange idéal :


avec :
- la pression de bulle du mélange liquide à ;
- la pression de rosée du mélange gazeux à ;
- l'enthalpie de vaporisation du corps pur à ;
- et les fractions molaires respectives du corps en phases liquide et gaz.



Puisque les enthalpies de vaporisation des corps purs sont des grandeurs positives, les deux pressions et idéales ont une pente négative en fonction de : elles ne peuvent donc qu'augmenter en fonction de . Or autour du point critique les courbes réelles présentent localement des pentes négatives en fonction de , voir figure 5. Par conséquent les courbes idéales ne peuvent pas représenter un point critique.

Pour un corps pur la formule de Clausius-Clapeyron donne :

avec la pression de vapeur saturante du composant à la température , d'où :



Un équilibre liquide-vapeur idéal suit la loi de Raoult. Pour chaque composant on a . En conséquence :



La loi de Raoult donne également et . En introduisant ces expressions dans les deux relations ci-dessus celles-ci deviennent explicites en la pression considérée et ne dépendent plus que de la température et de la composition de phase constante. D'autre part, en dérivant ces expressions on retrouve les relations ci-dessus : la formule de Clausius-Clapeyron étendue aux équilibres liquide-vapeur de mélanges idéaux est cohérente avec la loi de Raoult.


Notes et références
Notes
- Deux autres relations liées aux coefficients calorimétriques , le coefficient de dilatation isotherme, et , le coefficient de compression isotherme, sont également appelées relations de Clapeyron.
- É. Clapeyron, « Mémoire sur la puissance motrice de la chaleur », Journal de l'École polytechnique, vol. 23, , p. 153-191 (lire en ligne), p. 173.
- (de) R. Clausius, « Ueber die bewegende Kraft der Wärme und die Gesetze, welche sich daraus für die Wärmelehre selbst ableiten lassen » [« On the motive power of heat and the laws which can be deduced therefrom regarding the theory of heat »], Annalen der Physik, vol. 155, , p. 500–524 (DOI 10.1002/andp.18501550403, Bibcode 1850AnP...155..500C, lire en ligne), p. 505.
- Vidal 1997, p. 37.
- Vidal 1997, p. 42.
- Par exemple, dans les conditions normales de température et de pression, le volume molaire de l'eau liquide est voisin de 18 ml, celui de l'eau gaz de 22 400 ml.
- Vidal 1997, p. 38.
- Febvre et al., p. 575.
- Jacques Schwarzentruber, EMAC, « Expression de la pression de saturation liquide-vapeur », sur École nationale supérieure des mines d'Albi-Carmaux (consulté le ).
- Jean-Noël Jaubert et Louis Schuffenecker, Pressions de vapeur saturantes des composés organiques, vol. K 670, Techniques de l'ingénieur, (lire en ligne).
- « Vrai ou faux ? L'eau bout à 100 °C », sur L'Internaute.
- Jacques Schwarzentruber, EMAC, « Fusion de la glace par compression », sur École nationale supérieure des mines d'Albi-Carmaux.
- « Le regel de la glace (1990) - vidéo », sur Canal-U.
- Météo-France, « Phase », sur meteofrance.fr (consulté le ).
- Vidal 1997, p. 190-191.
- (en) Luh C. Tao, « Clapeyron Equation of a Multicomponent Solution », AlChE Journal, , p. 460 (lire en ligne, consulté le ).
- (en) James C. M. Li, « Clapeyron Equation for Multicomponent Systems », J. Chem. Phys., vol. 25, no 3, , p. 572-574 (lire en ligne, consulté le ).


Bibliographie
- Jean Vidal, Thermodynamique : application au génie chimique et à l'industrie pétrolière, Paris, Éditions Technip, coll. « Publications de l'Institut français du pétrole. », , 500 p. (ISBN 978-2-710-80715-5, OCLC 300489419, lire en ligne), p. 37-38, 42, 190-191.
- Jean-Pierre Corriou, Thermodynamique chimique : Définitions et relations fondamentales, vol. J 1025, Techniques de l'ingénieur, coll. « base documentaire Thermodynamique et cinétique chimique, pack Opérations unitaires. Génie de la réaction chimique, univers Procédés chimie - bio - agro », (lire en ligne), p. 1-19.
- Jean-Pierre Corriou, Thermodynamique chimique : Diagrammes thermodynamiques, vol. J 1026, Techniques de l'ingénieur, coll. « base documentaire Thermodynamique et cinétique chimique, pack Opérations unitaires. Génie de la réaction chimique, univers Procédés chimie - bio - agro », , p. 1-30.
- Pascal Febvre, Richard Taillet et Loïc Villain, Dictionnaire de physique, (ISBN 978-2-8041-7554-2), p. 227-228-693-575.
- Peter William Atkins et Julio De Paula, Chimie physique, De Boeck Supérieur, (ISBN 978-2-8041-6651-9), p. 146-147-148-149-150-151.
- Hervé Lemarchand, sous la direction de Geneviève M. L. Dumas et Roger I. Ben-Aïm, L'indispensable en thermodynamique chimique : les fondements, Bréal, (ISBN 2-7495-0264-0, lire en ligne), p. 72-75.
Liens externes
- Jacques Schwarzentruber, EMAC, « Relation de Clapeyron », sur École nationale supérieure des mines d'Albi-Carmaux.
Voir aussi
- Changement d'état
- Diagramme de phase
- Enthalpie de changement d'état
- Formule de Clausius-Clapeyron
- Formules d'Ehrenfest
- Relations de Clapeyron
- Transition de phase
 Portail de la physique
Portail de la physique  Portail de la météorologie
Portail de la météorologie