Sclérodermie
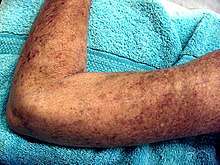
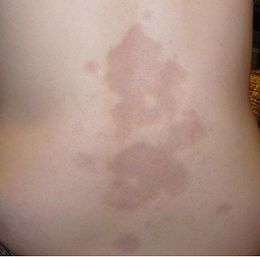
Les sclérodermies sont un « groupe hétérogène de maladies du tissu conjonctif d'étiologie inconnue qui ont en commun une induration et un changement d'aspect de la peau ». Les sclérodermies uniquement cutanées (dites « localisées ») se distinguent par des lésions scléreuses en formes de taches ou parfois grossièrement de bandes (siégeant alors plutôt sur les membres, le cuir chevelu et le visage). Ces lésions sont aussi appelées « morphées », par opposition aux sclérodermies systémiques qui affectent aussi les organes internes.
Chez la majorité des patients atteints de sclérodermie localisée, l'IRM révèle souvent des « anomalies de signal » indiquant des troubles musculosquelettiques[1] et même chez des patients asymptomatiques[1]. Ces troubles varient selon le sous type clinique[1].
| Spécialité | Immunologie |
|---|
| CIM-10 | L94.0-L94.1, M34 |
|---|---|
| CIM-9 | 701.0 710.1 |
| MedlinePlus | 000429 |
| MeSH | D012594 |
![]()
Les sclérodermies ne semblent pas contagieuses. Elles pourraient avoir une composante génétique car quelques cas "familiaux" sont rapportés par la littérature[2]. « Il y a de fortes présomptions que plusieurs gènes soient impliqués et puissent provoquer la maladie à la faveur de conditions environnementales, chimiques ou virales[2]. » Certains métiers semblent prédisposer à cette maladie (certains ouvriers de l'industrie, mineurs, sculpteurs)[3] ce qui invite à évoquer un lien avec l'inhalation de silice libre[4]. En 1753, la maladie fut décrite pour la première fois par le docteur de l'Hôpital des Incurables de Naples, Carlo Curzio[5].
Mécanismes et description
Les sclérodermies sont des maladies qui semblent liées à une accumulation du collagène et à un épaississement de la matrice extracellulaire, ce qui provoque une atteinte des vaisseaux de petit calibre qui deviennent fibreux au niveau de leur paroi. Plus profondément dans le réseau capillaire on constate une fibrose progressive accompagnée d'une ischémie. « Une activation endothéliale pourrait être à l’origine de ce processus avec augmentation de l’expression de molécules d’adhésion endothéliales (VCAM-1, ICAM-1 et selectine E) et recrutement de lymphocytes qui produisent des cytokines pro-fibrosantes (TGF-β, IL-4 et IL-6) et pro-inflammatoires. Le TGF-β augmente la production de collagène, de fibronectine et de protéoglycanes par les myofibroblastes, et diminue la production de protéases (MMP1 et MMP9), qui sont responsables de la dégradation du collagène ».
Il peut s'agir de maladies auto-immunes qui, dans leurs formes localisées, provoquent des indurations cutanées et une modification de couleur et d'aspect de la peau. Les petites lésions (en gouttes ou en plaques) peuvent régresser spontanément après quelques années sans séquelles graves.
La lésion s'étend parfois de manière linéaire (sclérodermie en bande ou sclérodermie linéaire[6]). les bandes sont généralement assez bien délimitées, formant un tissu ferme (difficile ou impossible à pincer) de couleur claire à blanc nacré, bordées par un liseré de couleur pourpre chez les individus à peau claire. La lésion peut aussi prendre l'aspect d'un coup de sabre qui aurait déformé un visage (dépression/enfoncement de la peau le long de la lésion). Quand la maladie apparait précocement, elle peut déformer les membres, les doigts et/ou la main[7] ou le crâne (avec amincissement de la boîte crânienne sous les lésions[8] et le visage[9],[10].
Les changements de couleur et texture de la peau et de structures sous-jacentes peuvent dans les cas de sclérodermie frontopariétales suivre sur le corps les lignes de Blaschko[11].
Typologie
Forme localisée et cutanée
Elle est caractérisée par
- une atteinte clinique apparemment limitée à la peau, avec des lésions qui peuvent être planes ou déprimées ;
- l'absence de sclérodactylie ;
- l'absence de syndrome de Raynaud ;
- l'absence d’atteinte microvasculaire (à la capillaroscopie).
Les organes internes ne sont pas touchés, sauf dans le cas de la scléro-atrophie de Degos qui est classée parmi les sclérodermies localisées, mais touche néanmoins également souvent le système musculaire des membres[12]. Cette forme est assez fréquente chez l'enfant et touche plutôt les membres inférieurs[12].
Forme CREST
C'est une forme limitée de sclérodermie, CREST reprenant les initiales des quatre pathologies associées :
- calcinose (C) : de petites masses calcifiées apparaissent sur la peau, avec parfois des ulcères en regard des saillies osseuses des pieds et mains ;
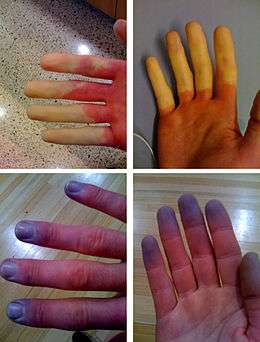
- Syndrome de Raynaud (R) : les artérioles des doigts (de mains/pieds) se contractent au froid, entraînant une chute de la vascularisation (ischémie digitale paroxystique, associée à une succession de pâleur et de cyanose (⇒ coloration gris-bleuté des doigts). Inversement un réchauffement trop brusque induit une coloration rouge vif des doigts avec une douleur. Parfois, seuls deux ou trois doigts de chaque main sont affectés par ce syndrome (qui peut aussi annoncer - mais pas systématiquement - une sclérodermie généralisée ou systémique ;
- Trouble œsophagique (E) : un déficit de péristaltisme nuit à la digestion. Il est mesuré par manométrie œsophagienne (prise de la pression à l’intérieur de l’œsophage) et se traduit par une dysphagie (gène quand le patient avale les aliments) ;
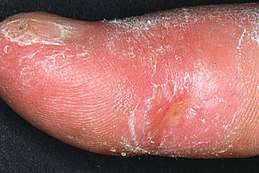
- Une sclérodactylie (S) : rigidification de la peau qui peut prendre l'aspect d'un parchemin impossible à pincer. Les doigts deviennent plus fins puis se déforment (flexion irréductible), alors qu'une télangiectasie peut aussi toucher le visage, les mains ou les pieds.
- Télangiectasies (T)
Forme généralisée
Elle est dite « généralisée » ou « systémique » (sclérodermie systémique) Elle peut attaquer de nombreux autres organes que la peau et notamment le cœur (coronaropathie), les poumons, le rein (néphropathie vasculaire) ou encore le tractus gastro-intestinal ; dans ces derniers cas le pronostic vital est en jeu, car aucun traitement efficace n'existe actuellement.
Formes chéloïdiennes (ou nodulaires)
Cela est rare, mais dans les formes cutanées localisées, comme dans les formes systémiques, des éléments nodulaires peuvent apparaître alors que la maladie évolue ; avec ou sans bandes scléreuses, avec un aspect atypique « bigarré » (zones pigmentées alternant avec des zones dépigmentées, avec la présence de nodules, observées chez des hommes adultes[13]. De tels nodules ont été signalés dans la littérature médicale dès 1884 (par Addisson). Ils peuvent parfois se manifester dès le début de la manifestation de la dermatose[13]. Dans ces cas, la sclérodermie est dite « chéloïdienne » ou « nodulaires ».
Autres formes frontières
- Les connectivites mixtes : elles associent des signes de sclérodermie, de lupus érythémateux disséminé et de dermatomyosite.
Le syndrome de Sharp en est l'expression la plus simple, incluant un syndrome de Raynaud, d'arthralgie des mains, des doigts d'apparence « boudinée », des mégacapillaires, et des anticorps antiribonucléoprotéine ; - le syndrome de Shulman ; c'est une fasciite qui infiltre le derme profond et les aponévroses, avec rétraction tendineuse progressive et d'éosinophilie, mais sans syndrome de Raynaud et sans aucune manifestation viscérale. Il conduit parfois à une aplasie médullaire grave ;
- la « sclérodermie du diabétique » : elle est caractérisée par l'évolution chronique et bénigne d'infiltrations du cou et des membres, très souvent œdémateuses et parfois dyschromiques ;
- le MMTC, syndrome de Cléopâtre et autres syndromes sclérodermiformes d'origine médicamenteuse et/ou toxique : c'est un syndrome toxicologique invalidant proche de la sclérodermie ; il est caractérisé par des fortes douleurs musculaires associées à une éosinophilie (taux anormal de globules blancs et des sclérodermies. Il affecte à la fois les muscles et le squelette.
Les empoisonnements peuvent être d'origine médicamenteuse[14] avec par exemple une sclérose cutanée au site d’injection de vitamine K1 par voie intramusculaire[15], ou de pentazocine, ou plus rarement de véritables sclérodermies systémiques[16],[17],[18],[19],[20].
Les empoisonnements peuvent aussi être environnementaux[18] et notamment d'origine alimentaire ; le cas le plus massif et connu est celui du syndrome de l'huile toxique SHT à Madrid en 1981, catastrophe sanitaire qui en raison de la diffusion d'une huile frelatée vendue comme huile alimentaire a fait environ 20 688 victimes dont — suivant les décomptes — de 370 à 835 sont mortes[21],[22]. Un autre exemple connu est celui du syndrome éosinophilie–myalgie lié au L-tryptophane et on a plus récemment démontré l'existence d'une dermopathie fibrosante néphrogénique induite par le gadolinium notamment utilisé comme agent de contraste pour la radiographie, en particulier chez des patients ayant une insuffisance rénale chronique. Il est à craindre que les patients victimes de fuites d'implants de silicone puissent aussi être victimes de troubles iatrogènes de ce type[23],[24],[25],[26],[27],[28],[29],[30],[31],[32],[33]. - « syndrome de chevauchement » (épaississement de la peau associé à une faiblesse musculaire) comme la sclérodermatomyosite (pouvant être confondue avec une polymyosite.
Associations pathologiques
Ce sont (en lien avec le syndrome de Reynolds)
Prévalence
La maladie est rare : elle touche 2 à 16 personnes sur un million, des femmes le plus souvent.
Les premiers symptômes apparaissent souvent entre 40 et 50 ans[2], mais les formes auto-immunes peuvent être contractées à tout âge.
La sclérodermie auto-immune atteint 3 femmes pour 1 homme et se déclare habituellement entre 30 et 50 ans.
Évolution
Elle est imprévisible et il n'existe pas de médicaments, si ce n'est pour traiter certains symptômes.
- Dans les formes localisées, la sclérodermie détruit définitivement les poils ou cheveux situés au niveau des lésions en plaques ou bandes. La sensibilité superficielle est dégradée et la sécrétion sudorale diminue ou disparaît également. Les taches peuvent s'étendre (en surface ou en nombre) ou se stabiliser ou disparaitre avec toutefois une « atrophie séquellaire secondaire (pigmentée ou dépigmentée) », avec perte de pilosité)[12].
- Dans les formes généralisées, le pronostic vital est en jeu.
Diagnostic
C'est une maladie dont les débuts sont difficiles à diagnostiquer, et qui peut mettre plusieurs années à se déclarer. Une biopsie cutanée précise le diagnostic.
Elle est généralement précédée de troubles conjonctifs bénins, et souvent associée à des troubles dépressifs[34].
Dans certains cas, les signes cliniques peuvent rapidement faire évoquer un syndrome de CREST à savoir
- calcinose sous-cutanée,
- phénomène de Raynaud (changement de coloration des doigts en contact avec le froid),
- troubles de la motilité œsophagienne avec dysphagie,
- sclérodactylie
- télangiectasies.
La biologie retrouve habituellement des anticorps anti-nucléaires et, de façon plus spécifique mais moins fréquente, des anticorps anti-Scl70.
Risque de cancer associé
Il semble exister. L'association de sclérodermie et de néoplasie est rare mais possible ; leur « caractère fortuit ou non reste controversé ». Il convient de rechercher dans les formes atypiques ou rapidement progressives une néoplasie sous-jacente.
Remarque : dans un cas, une morphée généralisée a complètement régressé deux mois après l’intervention chirurgicale d'un cancer de l'anus[35].
Traitement
Les traitements proposés à ce jour sont les corticoïdes, les immunosuppresseurs (cyclophosphamide : Endoxan, ou azathioprine (Imurel) et le traitement d'une éventuelle néoplasie sous-jacente.
Des prostaglandines (iloprost : Ilomédine) sont également proposées pour leur effet vasodilatateur qui permet parfois une amélioration significative.
Les corticoïdes et anti-inflammatoire non stéroïdien (AINS) agissent sur la composante douloureuse surtout articulaire ; les topiques ![]()
Notes et références
- (en) Qtefan Schanz, Gerhard Fierlbeck, Anja Ulmer, Marc Schmalzing, Jasmin Kümmerle-Deschner, ClausD Claussen, marius Horger (2011) « Localized scleroderma : MR Findings and Clinical features » Radiology vol260, no 3, septembre 2011, PDF, 17 p.
- site de l'Association romande des sclerodermiques, consulté 2013-10-05
- Association Sclérodermie (France) Qui est atteint par la Sclérodermie ?, consulté 2013-10-05
- Amoudru, C. (1991) « Sclérodermie généralisée et inhalation de poussières mixtes contenant de la silice libre » Documents pour le Médecin du Travail, 46, 101-106.
- http://www.asmn.re.it/lezione-magistrale-a-palazzo-rocca-saporiti-la-sclerodermia-una-malattia-ancora-misteriosa
- (en) Cure Bytes, « What is Linear Scleroderma? », consulté 2013-10-05
- Images.rheumatology.org Rheumatic Diseases of Childhood ; Linear Scleroderma: Hand , consulté 2013-10-05
- Images.rheumatology.org, Rheumatic Diseases of Childhood ; Linear Scleroderma: Facial Hemiatrophy , consulté 2013-10-05
- Images.rheumatology.org Rheumatic Diseases of Childhood > Localized Scleroderma: En Coup De Sabre, Face , consulté 2013-10-05
- Images.rheumatology.org Juvenile Localized Scleroderma: En Coup De Sabre and Morphea, Head and Trunk , consulté 2013-10-05
- Soma, Y., & Fujimoto, M. (1998). Frontoparietal scleroderma (en coup de sabre) following Blaschko's lines. Journal of the American Academy of Dermatology, 38(2), 366-368 (résumé).
- Association Sclérodermie « La Sclérodermie localisée » consulté 2013-10-05
- Bayle, P., Bazex, J., Marguery, M. C., & Lamant, L. (2005) « Nodules sur sclérodermie en plaque ou morphée » Annales de dermatologie et de vénéréologie (fév 2005, Vol. 132, No 2, p. 130-132) (résumé)
- (en) Bannwarth B. « Drug-induced musculoskeletal disorders » Drug Saf. 2007 ; 30 : 27-46
- (en) Pang BK, Munro V, Kossard S. « Pseudoscleroderma secondary to phytomenadione (Vitamin K1) injections: Texier’s disease » Austral J Dermatol. 1996 ; 37 : 44-7.
- (en) Haustein UF. « Scleroderma and pseudoscleroderma : uncommon presentations » Clin Dermatol. 2005 ; 23 : 480-90.
- (en) Haustein UF, Haupt B. « Drug-induced scleroderma and sclerodermiform conditions » Clin Dermatol. 1998 ; 16 : 353-66.
- (en) D’Cruz D. « Autoimmune diseases associated with drugs, chemicals and environmental factors » Toxicol Letters 2000 ; 112-113 : 421-32
- Hallé O, Schaeverbeke T, Bannwarth B, Dehais J. « Les facteurs d’environnement et les éléments iatrogènes dans la sclérodermie systémique et les syndromes apparentés. Revue de la littérature » Rev Méd Interne 1997 ; 18 : 219-29
- (en) Jablonska S, Blaszcyk M. « Scleroderma-like disorders » Semin Cutan Med Surg. 1998 ; 17 : 65-76
- Isabelle Marie « Sclérodermie systémique et syndromes sclérodermiformes d’origine médicamenteuse et toxique, et syndrome de Cléopâtre » Médecine thérapeutique Volume 14, Numéro 4, 226-36, juillet-août 2008, Dossier
- D’après (en) (en) Dorland's Medical Dictionary, (réimpr. 2003 (30e éd.)), 2224 p. (ISBN 0721601464), « Spanish toxic oil syndrome » qui donne plus de 600 morts et plus de 20 000 victimes
- (en) Dugowson CE, Daling J, Koepsell TD, Voigt L, Nelson JL. « Silicone breast transplants and risk for rheumatoid arthritis » Arthritis Rheum. 1992 ; 35(suppl) : S66.
- (en) Englert H, Small-McMahon J, Davis K, O’Connor H, Chambers P, Brooks P. « Male systemic sclerosis and occupational silica exposure – a population-based study » Aust N Z J Med. 2000 ; 30 : 215-20
- (en) Goldman JA, Greenblatt J, Joines R, White L, Aylward B, Lamm SH. « Breast implants, rheumatoid arthritis and connective tissue diseases in a clinical practice » J Clin Epidemiol. 1995 ; 48 : 571-82.
- (en) Hennekens CH, Lee IM, Cook NR. et al. « Self-reported breast implants and connective-tissue diseases in female health professionals » JAMA 1996 ; 275 : 616-21.
- (en) Hochberg MC, Perlmutter DL, Medsger Jr. TA. et al. « Lack of association between augmentation mammoplasty and systemic sclerosis (scleroderma) » Arthritis Rheum. 1996 ; 39 : 1125-31.
- (en) Janowsky EC, Kupper LL, Hulka BS. « Meta-analyses of the relation between silicone breast implants and the risk of connective-tissue diseases » N Engl J Med. 2000 ; 427 : 781-90.
- (en) Kjoller K, Friis S, Mellemkjoer L. et al. « Connective tissue disease and other rheumatic conditions following cosmetic breast implantation in Denmark » Arch Intern Med. 2001 ; 161 : 973-9.
- (en) Laing TJ, Gillepsie BW, Lacey Jr. JV. et al. « The association between silicone exposure and undifferentiated connective tissue disease among women in Michigan and Ohio » Arthritis Rheum. 1996 ; 39(suppl) : S150.
- (en) Lipworth L, Tarone RE, McLaughlin JK. « Silicone breast implants and connective tissue disease. An updated review of the epidemiologic evidence » Ann Plast Surg. 2004 ; 52 : 598-601.
- (en) Nyren O, Yin L, Josefsson S, McLaughlin JK. et al. « Risk of connective tissue disease and related disorders among women with breast implants: a nation-wide retrospective cohort study in Sweden » Br Med J. 1998 ; 316 : 417-22.
- (en) Wolfe P. « Silicone breast implants and the risk of fibromyalgia and rheumatoid arthritis » Arthritis Rheum. 1995 ; 38(suppl) : S265.
- (en) Roca RP, Wigley FM, White B. « Depressive symptoms associated with scleroderma » Arthritis Rheum. 1996 ; 39 (6) : 1035-1040.
- A. Masmoudi, M. Amouri, A. Khabir, A. Charfeddine, H. Ben Salah, M. Sallemi, A. Amouri, S. Boudaya, S. Bouassida and T. Boudawara et al. « Cas Clinique : Morphée généralisée révélatrice d’un adénocarcinome rectal (Generalized Morphea revealing rectal adenocarcinoma) » Oncologie Volume 10, Number 11 (2008), 673-676 ; DOI:10.1007/s10269-007-0790-2 (résumé)
Voir aussi
Articles connexes
Liens externes
- Page spécifique sur Orphanet
- Photographies de sclérodermies (atlas-dermato.org)
