Maladie de Minkowski-Chauffard
La maladie de Minkowski-Chauffard ou microsphérocytose héréditaire ou sphérocytose héréditaire est une maladie génétique, caractérisée par une anomalie des protéines constituant la membrane des globules rouges qui deviennent sphériques et fragiles, avec destruction intrasplénique, entrainant une anémie hémolytique chronique, un subictère et une splénomégalie.
| Maladie de Minkowski-Chauffard | |
| Référence MIM | 182900 |
|---|---|
| Transmission | Dominante |
| Chromosome | 8p11.2 |
| Liste des maladies génétiques à gène identifié | |
Épidémiologie
Il s'agit de la cause la plus fréquente d'hémolyse chronique héréditaire dans les pays occidentaux[1]. Sa prévalence est d'environ un cas pour 2 000[1].
Cause
Il s'agit d'une maladie héréditaire, et les deux sexes sont également affectés. La transmission est de type autosomique dominante par atteinte d'un gène réglant la synthèse d'une protéine de la membrane des globules rouges (ankyrine). Si le gène de la spectrine est muté, la maladie est autosomique récessive. La mutation d'un allèle ne suffit pas à provoquer l'apparition des symptômes associés à la maladie.
Pathogénie
Le défaut essentiel réside dans la forme anormale des globules rouges, qui sont des microsphérocytes. D'un diamètre globulaire trop petit, mais aussi d'une épaisseur trop grande, ces cellules ont une surface trop petite pour leur volume et sont dès lors très vulnérables à l'hémolyse osmotique : le moindre abaissement de la pression osmotique du milieu extérieur les fait éclater.
Cette anomalie morphologique doit reconnaître une base biochimique, peut-être enzymatique, mais la nature du défaut primaire reste inconnue. On sait seulement que les globules rouges normaux, exposés à un agent hémolysant (hémolysines + complément) et sur le point d'éclater, prennent également la forme sphérocytaire. Il est clair aussi que dans la sphérocytose héréditaire seuls les globules rouges d'un certain âge montrent cette anomalie : les réticulocytes et les globules rouges jeunes paraissent normaux. Il pourrait donc s'agir d'un vieillissement globulaire prématuré.
La maladie de Minkovski-Chauffard pourrait être liée à une spectrine (protéine membranaire du globule rouge) anormale. Cette protéine est un dimère associant une chaine α et une chaine β enroulées en double hélice. Ce dimère est relié de façon mobile à l'ankyrine, elle-même reliée à la bande III (deux autres protéines membranaires). Cet ancrage assure la déformabilité du globule rouge, lui conférant sa capacité de se faufiler dans des capillaires dont le diamètre est inférieur au volume globulaire. Une spectrine anormale est incapable de se fixer à l'ankyrine, provoquant l'apparition de cette maladie.
La rate, qui est toujours augmentée de volume (splénomégalie), a jadis été incriminée comme principale responsable de l'hyperhémolyse. Il est vrai que c'est elle qui a la fonction de détruire les sphérocytes, mais son augmentation de taille paraît secondaire au surcroît de travail qui lui incombe. La splénomégalie à son tour entraîne une hyperhémolyse, à laquelle peut mettre fin l'ablation chrirugicale de cet organe (splénectomie).
Symptômes cliniques
- L'ictère est le symptôme dominant : le malade est plus jaune qu'anémique. Son intensité varie d'un jour à l'autre, et il peut manquer par moments, ou pendant de longues périodes chez les sujets dont le foie épure très bien la bilirubine.
- La splénomégalie est évidente dans 70 % des cas. D'habitude modérée, elle augmente pendant les poussées d'hémolyse ; dans ce cas la rate devient spontanément très douloureuse et à la pression.
- Le foie est parfois augmenté de volume et la région vésiculaire peut devenir a son tour le siège de fortes douleurs spontanées ou provoquées.
- L'anémie est minime dans la majorité des cas et ne compromet généralement pas l'activité professionnelle du sujet. L'anémie est toutefois sujette à des poussées d'aggravation (voir complications).
- Des ulcères des jambes constituent un symptôme assez exceptionnel. Il existe parfois des taches pigmentaires au niveau des malléoles.
- Certaines anomalies de développement du squelette, et notamment le « crâne en forme de tour » se rencontrent peu fréquemment.
Examens de laboratoire
Examen hématologique
Mesures courantes
Le nombre de globules rouges est à peine réduit, sauf en période de poussée. Le taux d'hémoglobine indique que l'anémie est normochrome ou légèrement hyperchrome. Le volume globulaire moyen est soit normal, soit légèrement diminué (77 - 87 microns3). Si la réticulocytose est très élevée, ce volume peut même être accru. La concentration moyenne en hémoglobine globulaire est d'habitude un peu supérieure à la normale (37 % au lieu de 34 %).
Examen du frottis sanguin
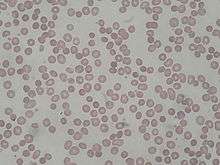
La microsphérocytose frappe immédiatement : beaucoup de globules rouges (mais non tous) ont un diamètre globulaire très petit (6,0 - 7,2 microns au lieu de 7,5) et sont intensément colorés, sans présenter l'éclaircissement central habituel (à cause de la perte de la forme biconcave). Comme les réticulocytes et les hématies les plus jeunes ne présentent pas cette anomalie, la distribution des diamètres globulaires montre de l'anisocytose, définie par une grande variation de taille.
La réticulocytose intense (20 - 30 %) est le second trait caractéristique du frottis. Quelquefois on aperçoit l'un ou l'autre érythroblaste. Il n'y a pas de poïkilocytose sauf dans les formes sévères.
Résistance globulaire osmotique
La résistance osmotique est extrêmement abaissée. La courbe d'hémolyse se déroule pour une concentration en sodium comprise entre 0,50 et 0,70 % de (normalement entre 0,40 et 0,45 % de NaCl). C'est là le trait le plus caractéristique de l'affection et il a une grande valeur diagnostique mais sa sensibilité n'est pas optimale, surtout dans les formes asymptomatiques[2]. L'autohémolyse à 37 °C est accrue.
Réaction de Coombs
La réaction de Coombs directe (RCD), appelée également test direct à l'antiglobuline (TDA), à la recherche d'auto-anticorps, est négative.
Globules blancs
Les globules blancs (ou leucocytes) sont en nombre normal ou légèrement augmenté. Pendant les crises d'hyperhémolyse il existe souvent une certaine leucopénie (baisse du nombre de globules blancs), qui fait place à de l'hyperleucocytose neutrophile pendant les phases de régénération.
Examen de la moelle osseuse
Elle est fortement érythroblastique comme dans toutes les anémies hémolytiques. Cet examen est inutile pour le diagnostic de la maladie.
Données métaboliques
L'hyperbilirubinémie et l'urobilinurie sont celles de tout ictère hémolytique. L'haptoglobine sérique est d'habitude absente ou très abaissée. L'hyperhémolyse n'est généralement pas assez aiguë pour donner lieu à une hémoglobinurie.
Évolution et complications
La sphérocytose héréditaire est en général une maladie bénigne compatible avec une espérance de vie à peu près normale. L'ictère est fluctuant et s'aggrave dans certaines circonstances, notamment à l'occasion de la grossesse, du surmenage physique, de l'exposition au froid et des émotions. Dans un cinquième de cas, la maladie peut ne présenter aucun signe et n'être découverte que lors d'un bilan systématique[1].
Le malade est toutefois exposé à certaines complications :
- des crises d'hyperhémolyse grave peuvent peuvent survenir spontanément ou à l'occasion des circonstances énumérées plus haut. Les symptômes sont ceux de toute crise hémolytique (frissons, haute température, dyspnée, palpitations, douleur et gonflement de la rate et du foie, chutes importantes des globules rouges qui peuvent tomber en dessous d'un million par millimètre-cube). L'une des crises peut devenir fatale, mais la régénération est habituelle ;
- le blocage médullaire est caractérisé par l'aggravation de l'anémie, la rétrocession de l'ictère et la diminution de la réticulocytose. Cette complication est potentiellement fatale. L'une des causes les plus fréquente est une infection à certains types de parvovirus[3] ;
- la lithiase biliaire par calculs pigmentaires est quasi inévitable et peut survenir dès l'enfance[4]. Elle peut rester latente mais souvent elle imposera la cholécystectomie (ablation chirurgicale de la vésicule bilaire) ;
- une insuffisance cardiaque peut survenir par suite de l'anémie chronique.
Traitement
La splénectomie (ablation chirurgicale de la rate) est le seul traitement valable. Elle ne supprime pas le défaut intrinsèque des globules rouges mais elle prolonge notablement la survie des sphérocytes. De ce fait, le pourcentage de ceux-ci dans le sang circulant s'accroît après l'opération. On verra également apparaître dans le frottis sanguin les stigmates habituels de l'absence de fonction splénique : la présence de sidérocytes et d'hématies à corps de Jolly. Les résultats cliniques sont excellents : les crises hémolytiques cessent en général de se produire, l'ictère diminue et l'anémie rétrocède, et souvent disparaît. On tiendra compte d'une possibilité de récidive par suite du développement vicariant de rates accessoires ou d'autres suppléances du système réticulo-endothélial. Les indications de la splénectomie doivent être larges lorsque la maladie compromet l'activité normale du sujet et sont formelles dès qu'il existe des complications. On aura intérêt à ne pas retarder l'intervention chez des sujets jeunes. La splénectomie permet, en outre, d'éviter la formation de lithiase biliaire[5].
On évitera ou espacera tant que possible les transfusions de sang et on considérera le fer comme nettement contre-indiqué.
Notes et références
- (en) Perrotta S, Gallagher PG, Mohandas N. « Hereditary spherocytosis » Lancet 2008;372:1411-26.
- (en) Korones D, Pearson HA, « Normal erythrocyte osmotic fragility in hereditary spherocytosis » J Pediatr. 1989;114:264-6.
- (en) Brown KE, « Haematological consequences of parvovirus B19 infection » Baillieres Best Pract Res Clin Haematol. 2000;13:245-59.
- (en) Tamary H, Aviner S, Freud E. « High incidence of early cholelithiasis detected by ultrasonography in children and young adults with hereditary spherocytosis » J Pediatr Hematol Oncol. 2003;25:952-4.
- (en) Sandler A, Winkel G, Kimura K, Soper R, « The role of prophylactic cholecystectomy during splenectomy in children with hereditary spherocytosis » J Pediatr Surg. 1999;34:1077-8.
Lien externe
- Sphérocytose héréditaire sur orpha.net

