Syndrome de Vogt-Koyanagi-Harada
Le syndrome de Vogt-Koyanagi-Harada, aussi appelé syndrome uvéoméningite et syndrome uvéoméningoencéphalitique, est une maladie auto-immune rare caractérisée par une attaque du système immunitaire contre les mélanocytes[1]. Les symptômes sont d'ordre oculaires, méningés, auditifs et cutanés[2]. La manifestation la plus significative est l'uvéite (inflammation) des deux yeux[3],[4]. Il peut aussi affecter l'oreille interne avec des effets sur l’ouïe, la peau et les méninges du système nerveux central[3],[4],[5],[6],[7].

| Spécialité | Ophtalmologie |
|---|
| eMedicine | 1229432 |
|---|---|
| MeSH | D014607 |
| Symptômes | Méningite |
![]() Mise en garde médicale
Mise en garde médicale
Signes et symptômes
Présentation
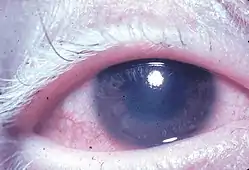
La maladie se caractérise par une uvéite diffuse, des douleurs, des rougeurs et un flou de la vision. Les symptômes oculaires peuvent être accompagnés de divers symptômes systémiques, tels que auditifs (acouphènes[7], vertiges[7] et de la surdité), neurologiques (syndrome méningé, avec malaises, fièvres, maux de tête, nausées, douleurs abdominales, raideur du cou et du dos, ou une combinaison de ces facteurs[7] ; méningite[5], pléocytose (en) du LCS, paralysie des nerfs crâniens, hémiparésie, myélite transverse et ganglion ciliaire[7]), et des manifestations cutanées, comme l'achromotrichie, le vitiligo, et l'alopécie[5],[6],[7]. Le vitiligo se retrouve souvent dans la région du sacrum[7].
Phases
La séquence des événements cliniques du Vogt-Koyanagi-Harada se divise en quatre phases : prodromique, uvéitique aiguë, convalescente et récurrente chronique[3],[6],[7].
La phase prodromique peut ne présenter aucun symptôme ou imiter une infection virale non spécifique, marquée par des symptômes pseudo-grippaux qui durent généralement quelques jours[7]. Il peut y avoir manifestation de fièvre, de maux de tête, de nausées, de syndrome méningé, de dysacousie (en) (inconfort causé par des bruits forts ou distorsion de la qualité des sons entendus), des acouphènes et/ou vertiges[7],[8]. Les symptômes oculaires peuvent comprendre des douleurs à l'orbite, une photophobie et des déchirures de la vision (en)[7]. La peau et les cheveux peuvent devenir sensibles au toucher[7],[8]. Une paralysie des nerfs crâniens et une névrite optique sont rares[7].
La phase uvéitique aiguë survient quelques jours plus tard et dure généralement plusieurs semaines[7]. Elle est annoncée par une panuvéite bilatérale provoquant un flou de la vision[7]. Dans 70% des cas, l'apparition de flous visuels a lieu aux deux yeux. S'il n'est que sur un seul au début, l'autre œil est touché en quelques jours[7]. Le processus peut inclure une uvéite antérieure granulomateuse aux yeux, un degré variable de vitrite, un épaississement de la choroïde postérieure avec élévation de la couche choroïdienne rétinienne péripapillaire, une hyperémie du nerf optique et une papillite optique (en), et des décollements multiples de rétine séreuse bulleuse exsudative[3],[6],[7].
La phase de convalescence se caractérise par une dépigmentation progressive des tissus de la peau avec un vitiligo et de l'achromotrichie, parfois avec des cicatrices numigulaires dépigmentées, ainsi que de l'alopécie et une dépigmentation du fond de l’œil qui se traduit par une décoloration orange-rouge classique (« fond coucher du soleil[6],[9],[8] ») et une agglutination et/ou migration de l'épithélium pigmentaire de la rétine[3],[7].
La phase récurrente chronique peut être marquée par des épisodes d'uvéite à répétition, mais est le plus souvent une uvéite chronique, de faible gravité, souvent subclinique, pouvant entraîner une inflammation antérieure des granulomes, des cataracts, du glaucome et une hypertension oculaire[3],[4],[6],[7]. Les récidives entière sont toutefois rares après la fin de la phase aiguë[9].Une dysacousie peut se produire durant cette phase[8].
Causes
Bien qu'il y ait parfois une infection virale ou un traumatisme cutané ou oculaire[7], l'initiateur sous-jacent exact de la maladie de Vogt-Koyanagi-Harada reste inconnu[5]. Cependant, elle est attribuée à une réponse immunitaire aberrante induite par les lymphocytes T dirigée contre les auto-antigènes trouvés sur les mélanocytes[4],[5],[7]. Stimulé par l'interleukine 23 (IL-23), les lymphocytes Th17 et les cytokines comme l'interleukine 17 (IL-17) semblent cibler les protéines dans le mélanocyte[9],[10].
Facteurs à risque
Les personnes touchées ont généralement entre 20 et 50 ans[4],[5]. Le ratio femme/homme est de 2 pour 1[5],[6],[7]. Par définition, il n’existe aucun antécédent chirurgical ou traumatisme oculaire accidentel pouvant l'expliquer[4]. Le syndrome de Vogt-Koyanagi-Harada est plus commun chez les Asiatiques, les Latinos, les Moyen-Orientaux, les Indiens d'Amérique et les Métis mexicains, et beaucoup moins fréquent chez les Caucasiens et chez les Noirs d'Afrique subsaharienne[4],[5],[6],[7].
Le syndrome de Vogt-Koyanagi-Harada est associé à divers polymorphismes génétiques liés à la fonction immunitaire, comme par exemple aux antigènes des leucocytes humains DR4 et DRB1/DQA1[11] à la variabilité du nombre de copies de la composante complémentaire 4 (en)[11], un locus variant de l'interleukine 23[11] et à divers autres gènes non-antigènes[11]. L'antigène DRB1*0405, en particulier, semble jouer un rôle de susceptibilité important[3],[5],[9],[7].
Diagnostiques
Si détecté durant la phase prodromique, la pléocytose (en) du LCS est retrouvée dans plus de 80% des cas[7],[8], principalement dans les lymphocytes[8]. Cette pléocytose se résout en 8 semaines environ, même si l’uvéite chronique persiste[8].
Les tests fonctionnels peuvent inclure l'électrorétinogramme et des tests de champ visuel (en)[3]. La confirmation du diagnostic et une estimation de la gravité de la maladie peuvent demander des tests d'imagerie tels que l'ophtalmoscopie, l'angiographie à la fluorescéine ou au vert d'indocyanine (en), la tomographie en cohérence optique et les ultrasons[3],[6],[10],[8]. Par exemple, une angiographie au vert d'indocyanine peut détecter une inflammation choroïdienne continue dans les yeux sans symptômes cliniques ni signes préalables[6],[9]. L'IRM oculaire peut être utile[7] et les symptômes auditifs doivent subir des tests[7]. Les observations histopathologiques des yeux et de la peau sont discutées par Walton[7].
Le diagnostic du Vogt-Koyanagi-Harada se base sur la présentation clinique. Le diagnostic différentiel est étendu et comprend (entre autres) l'ophtalmie sympathique (en), la sarcoïdose, le lymphome à cellules B (en) intraoculaire primaire, une sclérite postérieure, un syndrome d'épanchement de l'uvée, la tuberculose, la syphilis, et les syndromes de choroïdopathie multifocale[4],[7].
Types
Sur la base de la présence de détections ailleurs qu'aux yeux, telles que des manifestations neurologiques, auditives et tégumentaires, les « critères diagnostiques révisés » de 2001[3],[12] classent la maladie comme complète (yeux et troubles neurologiques et cutanés), incomplète (yeux et sensation neurologique ou cutanée) ou probable (yeux, mais ni neurologiques ni cutané)[13],[4],[6],[7],[12]. Par définition, pour des raisons d'homogénéité de la recherche, il existe deux critères d'exclusion : un traumatisme oculaire pénétrant antérieur ou une chirurgie antérieure, et une maladie oculaire concomitante similaire à la maladie de Vogt-Koyanagi-Harada[3],[7],[12].
Soin
La phase d'uvéite aiguëe du syndrome de Vogt-Koyanagi-Harada réagit généralement à une dose élevée de corticostéroïdes par voie orale mais l'administration parentérale n'est généralement pas nécessaire[3],[4],[7]. Cependant, les complications oculaires peuvent nécessiter une injection de corticostéroïdes ou de bévacizumab dans la capsule de Tenon[7] ou le corps vitré[5],[7],[10]. En cas de rejets, d'autres immunosuppresseurs tels que la ciclosporine[3],[4], ou le tacrolimus[10], les antimétabolites (azathioprine, acide mycophénolique ou méthotrexate[10]), ou des agents biologiques tels que l'immunoglobuline intraveineuse ou l'infliximab, peuvent être nécessaires[3],[7].
Pronostic
Un pronostic visuel est généralement bon pour un diagnostic rapide et un traitement immunomodulateur agressif[3],[4],[9]. Les symptômes de l’oreille interne répondent généralement à une corticothérapie en quelques semaines à quelques mois; l'ouïe récupère généralement complètement[7]. Des effets chroniques des yeux tels que la cataracte, le glaucome et la neuropathie optique peuvent arriver[7]. Les modifications cutanées persistent généralement malgré le traitement[7].
Éponyme
Le syndrome de Vogt-Koyanagi-Harada est nommé d'après l'ophtalmologiste suisse Alfred Vogt et les Japonais Yoshizo Koyanagi (en) et Einosuke Harada (en)[14],[15],[16],[17]. Plusieurs auteurs, dont le médecin arabe Mohamed ibn Aslam Al-Ghafiqi au XIIe siècle, ainsi que Jacobi, Edward Nettleship (en) et Warren Tay au XIXe siècle, ont décrit la poliose, les névralgies et les troubles auditifs[17]. Ces symptômes étaient probablement dues à de l'ophtalmie sympathique (en) mais incluaient probablement des exemples du syndrome de Vogt-Koyanagi-Harada[17]. La première description de la maladie faite par Koyanagi remonte à 1914, mais avait été précédée en 1911 par Jujiro Komoto, professeur d'ophtalmologie à l'université de Tokyo[17]. C'est un article postérieur, publié en 1929, qui associe définitivement Koyanagi à la maladie[17]. L'article de Harada datant de 1926 est reconnu pour sa description complète de ce que l'on appelle aujourd'hui la maladie de Vogt–Koyanagi–Harada[17].
Notes et références
- « Syndrome de Vogt-Koyanagi-Harada », sur doctissimo.fr (consulté le ).
- Alaouia FZ, Benamoura S, El Kablia H, Amraou A, Syndrome de Vogt-Koyanagi-Harada. À propos de huit cas, La Revue de médecine interne, 2007;28:250–254
- Sakata VM, da Silva FT, Hirata CE, de Carvalho JF, Yamamoto JH, « Diagnosis and classification of Vogt-Koyanagi-Harada disease », Autoimmun Rev, vol. 13, nos 4-5, , p. 550–5 (PMID 24440284, DOI 10.1016/j.autrev.2014.01.023)
- Damico FM, Kiss S, Young LH, « Vogt-Koyanagi-Harada disease », Semin Ophthalmol, vol. 20, no 3, , p. 183–90 (PMID 16282153, DOI 10.1080/08820530500232126)
- Greco A, Fusconi M, Gallo A, « Vogt-Koyanagi-Harada syndrome », Autoimmun Rev, vol. 12, no 11, , p. 1033–8 (PMID 23567866, DOI 10.1016/j.autrev.2013.01.004)
- Cunningham ET, Rathinam SR, Tugal-Tutkun I, Muccioli C, Zierhut M, « Vogt-Koyanagi-Harada disease », Ocul. Immunol. Inflamm., vol. 22, no 4, , p. 249–52 (PMID 25014114, DOI 10.3109/09273948.2014.939530, lire en ligne)
- Walton RC, « Vogt-Koyanagi-Harada Disease », sur Medscape,
- (en) PK Rao et NA Rao, Uveitis and Immunological Disorders, Berlin, Heidelberg, Springer Science & Business Media, coll. « Essentials in Ophthalmology », , 145–155 p. (ISBN 978-3-540-30798-3, lire en ligne), « Chapter 10. Vogt–Koyanagi–Harada disease and Sympathetic Ophthalmia »
- Damico FM, Bezerra FT, Silva GC, Gasparin F, Yamamoto JH, « New insights into Vogt-Koyanagi-Harada disease », Arq Bras Oftalmol, vol. 72, no 3, , p. 413–20 (PMID 19668980, DOI 10.1590/s0004-27492009000300028, lire en ligne)
- Bordaberry MF, « Vogt-Koyanagi-Harada disease: diagnosis and treatments update », Curr Opin Ophthalmol, vol. 21, no 6, , p. 430–5 (PMID 20829689, DOI 10.1097/ICU.0b013e32833eb78c)
- Hou S, Kijlstra A, Yang P, « Molecular Genetic Advances in Uveitis », Prog Mol Biol Transl Sci, vol. 134, , p. 283–298 (PMID 26310161, DOI 10.1016/bs.pmbts.2015.04.009)
- Read RW, Holland GN, Rao NA, « Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature », Am. J. Ophthalmol., vol. 131, no 5, , p. 647–52 (PMID 11336942, DOI 10.1016/s0002-9394(01)00925-4)
- « Vogt-Koyanagi-Harada Disease », sur National Organization for Rare Disorders,
- Vogt A. Frühzeitiges Ergrauen der Zilien und Bemerkungen über den sogenannten plötzlichen Eintritt dieser Veränderung. Klinische Monatsblätter für Augenheilkunde, Stuttgart, 1906, 44: 228-242.
- Koyanagi Y. Dysakusis, Alopecie und Poliosis bei schwerer Uveitis nicht traumatischen Ursprungs. Klinische Monatsblätter für Augenheilkunde, Stuttgart, 1929, 82: 194–211.
- Harada E. Clinical study of nonsuppurative choroiditis. A report of acute diffuse choroiditis. Acta Societatis ophthalmologicae Japonicae, 1926, 30: 356.
- Herbort CP, Mochizuki M, « Vogt-Koyanagi-Harada disease: inquiry into the genesis of a disease name in the historical context of Switzerland and Japan », Int Ophthalmol, vol. 27, nos 2-3, , p. 67–79 (PMID 17468832, DOI 10.1007/s10792-007-9083-4)
 Portail de la médecine
Portail de la médecine