Liaison halogène
La liaison halogène (XB) est une liaison de nature proche de la liaison hydrogène (HB). Elle s'établit entre les atomes d'halogènes électrodéficients (appauvris en densité électronique), facilement polarisables tels que l'iode (I) et le brome (Br), et les entités riches en densité électronique comme les dérivés azotés et oxygénés (amines, éthers, amides, etc). Les applications pratiques et potentielles de la liaison halogène sont aussi vastes que celles de son analogue, la liaison hydrogène.
Définition
Une liaison halogène (XB) est une interaction électrostatique entre un halogène appauvri en électrons (polarisé δ+), c'est-à-dire un acide de Lewis, et une base de Lewis (polarisée δ−). L'appellation liaison halogène est introduite pour faire le parallèle avec la liaison hydrogène. Par ailleurs, les halogènes sont aussi connus pour former des liaisons covalentes avec les autres atomes comme le carbone, l'azote, l'oxygène, etc. Une liaison halogène (interaction halogène) fait essentiellement référence à une interaction non covalente où l'atome d'halogène agit comme entité électrophile. La liaison halogène est liée à la présence d'un trou sigma sur l'axe carbone-halogène due à la polarisabilité des atomes d'halogènes, en particulier l'iode et le brome.
Liaison

Analogie entre liaison halogène et liaison hydrogène
Une analogie existe entre la liaison halogène (XB) et la liaison hydrogène (HB)[1]. Dans les deux cas de liaisons, il s'agit d'une interaction entre un donneur d'électron et un accepteur d'électron. La différence entre les deux formes d'interaction réside dans la nature du site donneur. Dans le cas de la liaison hydrogène, l'hydrogène est l'accepteur d'électron et interagit de manière non-covalente en acceptant une densité électronique de la part d'une espèce riche en électron (exemple l'interaction O-H...O dans l'eau). Tandis que dans le cas de la liaison halogène, l'atome d'halogène est l'accepteur d'électron. Les transferts électroniques entre les deux espèces provoquent l'interpénétration des volumes de van der Waals[2].
Participants
Les études expérimentales et les considérations théoriques ont montré que les atomes d'halogènes qui peuvent participer la formation d'une liaison halogène sont : l'astate[3] (At), l'iode (I), le brome (Br), le chlore (Cl), et parfois le fluor (F). Tous les cinq sont capables de se comporter comme accepteur dans les liaisons halogènes (XB). La force de ces liaisons varie selon l'halogène engagé. Une tendance générale ressort, les halogènes les plus lourds tendent à donner lieu aux interaction les plus fortes : At > I > Br > Cl > F.
Les dihalogènes (I2, Br2, etc) ont tendance à former des liaisons halogènes fortes. Toutefois, la force et l'efficacité des interactions produites par le chlore et le fluor dépendent de la nature du donneur D. En effet, si l'halogène est lié à un groupement fortement électro-attracteur, il en résulte probablement des interactions plus fortes[4].
Caractéristiques
Les liaisons halogènes sont caractérisées par leur force directionnelle ce qui conduit des structures avec des architectures bien définies pouvant être raisonnablement prévues. La force de ces interactions varie entre 5 et 180 kJ/mol. Cette force des liaisons halogènes permet de la mettre en compétition avec les liaisons HB. Par ailleurs, les atomes D, X et A participant à la formation Halogène ont tendance à s'aligner et à former des angles se rapprochant le plus de 180°, ceci déjà démontré par Hassel[5] dans le cas du complexe entre le dibrome et 1,4-dioxane. Un autre facteur qui contribue à la force d'une liaison halogène est lié à la faible distance entre l'halogène (acide de Lewis, donneur XB) et la base de Lewis (accepteur XB). La nature attractive de l'interaction affecte la distance entre le donneur et l'accepteur qui est inférieure à la somme des rayons de van der Waals. La force de cette interaction augmente quand la distance entre l'halogène et l'accepteur diminue.
Historique

En 1863, Frederick Guthrie a rapporté pour la première fois la tendance qu'ont les halogènes à former des adduits avec les espèces électro-donneuses[6]. En effet, l'ajout de I2 à une solution saturée de nitrate d'ammonium NH4NO3, conduit à la formation de NH3I2. Ce dernier se décompose spontanément en ammoniac (NH3) et iode (I2) lorsqu'il est exposé à l'air. Ces constatations ont permis à Guthrie de conclure que le composé est de formule NH3I2 et qu'il s'agit d'un adduit obtenu par interaction non covalente et qu'il ne s'agit pas de la formation d'une liaison covalente N-I.
Grâce à la théorie de Robert S. Mulliken[7],[8], élaborée au milieu des années cinquante, sur les complexes obtenus par un effet de transfert d'électrons entre les donneurs et les accepteurs, une meilleure compréhension des mécanismes à travers lesquels les liaisons halogènes peuvent avoir lieu a été possible.
Mise en évidence
Les techniques spectroscopiques, la diffraction des rayons X (DRX), et les calculs théoriques ont permis de mieux comprendre la nature et les mécanismes des interactions halogènes.
par la DRX

La première preuve cristallographique de la formation de la liaison halogène a été apporté par Hassel en 1954. Il a pu élucider la structure de la dibromo-1,4-dioxanate par diffraction des rayons X[9]. Il a ainsi montré que la cohésion de la structure du cristal est assurée par des liaisons halogènes entre le brome et l'oxygène. La présence d'une interaction entre le brome et l'oxygène induit une diminution de la distance O-Br dans le cristal (2,71 Å) qui est de ~ 20 % inférieure à la somme des rayons de van der Waals de l'oxygène et du brome(3.35 Å). Un alignement des atomes participant à la liaison halogène est aussi constaté, en effet l'angle entre les liaisons O-Br et Br-Br est proche de 180°. Ceci a permis à Hassel de conclure que les atomes d'halogènes sont directement liés aux paires d'électrons n du donneur et que la direction de la liaison coïncide avec les axes des orbitales des doublets libres dans les composés donneurs[10].
par les calculs théoriques

Les calculs théoriques ab-initio permettent d'accéder à de nombreux paramètres électroniques au sein des édifices moléculaires halogénés tels que les charges partielles des atomes, la densité électronique, les distances interatomiques, la polarisabilité, etc. En effet, l'étude de la répartition de la densité électronique et les surfaces de potentiels électrostatiques des dérivés halogénés montrent l'existence d'une zone chargée positivement à la surface des halogènes fortement polarisables tels que l'iode (I) ou le brome (Br). Cette zone est appelée trou σ (sigma). L'apparition du trou σ peut être corrélée avec l'effet inductif attracteur qu'exercent les atomes ou les groupements d'atomes sur le nuage électronique de l'halogène en question. Ainsi, par exemple, l'étude de distribution de la densité électronique à la surface de l'iodobenzène et du pentafluoroiodobenzène en se fondant sur la théorie de la perturbation de Møller-Plesset du second ordre (MP2)[11] suivie par la cartographie de la répartition de la densité à la surface de ces molécules (surface de van der Waals ~ 0.002e/Å2) révèle une zone centrée autour de l'axe de la liaison C-I où l'iode est polarisé positivement (zone bleu) et le noyau aromatique a une forte densité électronique ainsi que les atomes de fluor (zone rouge). L'augmentation de l'effet attracteur par la substitution des hydrogènes par des fluors accentue et augmente la taille du trou σ.
Politlzer et coll[12]. ont montré que l'établissement de liaisons halogènes est intimement lié à la présence des trous σ à la surface des halogènes. La taille du trou σ est calculée en considérant la force de répulsion électrostatique obtenue entre la zone chargée positivement et une charge positive entière fictive. Elle est exprimée en kJ/mol.
par spectroscopie IR et RMN

Les études spectroscopiques notamment par la spectroscopie de rotation, la spectroscopie micro-onde à transformée de Fourier, la spectroscopie infrarouge à transformée de Fourier (FTIR), etc. ont fourni un bon nombre de preuves supplémentaires de la formation des liaisons halogènes[13]. Par exemple, la spectroscopie FTIR a permis de voir la présence des interactions XB à travers un léger changement de la fréquence de vibration des liaisons covalentes C-X[14]. Plus récemment, la RMN de l'azote 15 a été exploitée avec succès afin de mettre en évidence la présence de liaisons halogènes dans une molécule enrichie à l'azote N15 et contenant deux atomes d'iode[15]. Ainsi, la CP-MAS RMN (la RMN de l'état solide à polarisation croisée) a montré la présence de deux signaux relatifs à deux liaisons halogènes au sein du solide du fait que l'azote N15 interagit avec deux iodes situés dans deux positions cristallographiques différentes.
Applications
Les interactions halogènes ont servi pour l'élaboration de cristaux liquides, des polymères conducteurs, matériaux nanoporeux, etc. Par ailleurs, les liaisons halogènes gouvernent probablement la reconnaissance des médicaments ou des hormones halogénés par leurs sites de reconnaissance respectifs[16].
Ingénierie des cristaux
L'ingénierie des cristaux est un domaine récent de la recherche qui jette un pont entre la chimie de l'état solide et la chimie supramoléculaire[17]. Ce champ de recherche est par excellence interdisciplinaire car il fait appel à des disciplines classiques diverses telles que la cristallographie, la chimie organique et la chimie analytique. En 1971, Schmidt a initié ce champ en publiant un article sur la photodimérisation à l'état solide[18]. Une définition récente identifie l'ingénierie des cristaux comme l'exploitation des interactions intermoléculaires pour la cristallisation et le développement de nouveaux composés ayant des propriétés physicochimiques intéressantes. Avant la découverte de la liaison halogène, l'approche pour l'ingénierie des cristaux faisait appel à la liaison hydrogène, la chimie de coordination et les interactions inter-ionique pour le développement de matériaux à base de cristaux liquides ou de cristaux solides. De plus, la liaison halogène est utilisée pour organiser les sels des radicaux-cations, pour fabriquer des fils moléculaires conducteurs, et pour créer des édifices à base de cristaux liquides. De nouveaux matériaux à base de liaison halogène sont aussi apparus[19].
Cristaux liquides (LCs)

La nature particulière de l'interaction halogène peut servir comme nouvel outil pour élaborer des cristaux. Effectivement, en 2004, Loc Nguyen et al. ont été les premiers à exploiter les liaisons halogènes pour l'élaboration de cristaux liquides en utilisation comme précurseurs de départ alkoxystilbazoles et pentafluoroiodobenzene[20]. Par ailleurs, divers alkoxystilbazoles ont été utilisés pour la fabrication de matériaux pour l'optique non linéaire[21].
Resnati et coll. ont préparé des complexes à partir de l'iodopentafluorobenzene et des 4-alkoxystilbazoles. La DRX de ces complexes a révélé une courte distance N-I égale à 2.81 Å ce qui est inférieur à la somme des rayons de VdW (3.53 Å) tandis que l'angle B-X...A est égal à 168,4°. La faible distance de l'interaction N—I est en faveur d'une liaison très forte. Par ailleurs, la diffraction des rayons X ne montre pas d'interaction du type quadripolaire. L'utilisation du bromopentafluorobenzène comme acide de Lewis avec les 4-alkoxystilbazoles ne permet pas de former des complexes. Ces résultats ont permis de mettre en évidence l'effet de la polarisabilité de l'atome d'halogène.
Préparation de poly(diiododiacétylène)[22]

En l'an 2000, le prix Nobel de chimie fut décerné à Alan J. Heeger, Alan MacDiarmid, et Hideki Shirakawa qui ont découvert que le polyacétylène est un matériau conducteur et dont la conductivité est multipliée par un facteur 108 par oxydation avec le diiode (I2). Malgré la difficulté de le préparer, depuis lors un grand nombre de travaux ont essayé de mimer le squelette conjugué de ce polymère, par exemple le poly(p-phénylvinylène) à cause des nombreuses applications pratiques des polymères conjugués. En effet, ils sont utilisés pour l'élaboration des cellules photovoltaïques, diodes, transistor à effet de champ, capteurs chimiques, etc.
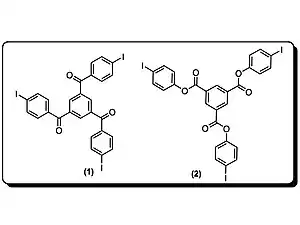
Goroff et al. ont préparé des poly(diiododiacétylène) ordonnés (PIDA) via le réarrangement du monomère (2) modulé par les liaisons halogènes. PIDA est excellent précurseur pour d'autres polymères conjugués, vue que l'iode peut être facilement échangé. Actuellement, la coupure de la liaison covalente C-I est possible par réduction électrochimique.
Les structures cristallines du monomère (2) sont celles de matériaux désordonnés de composition et connectivité variable.
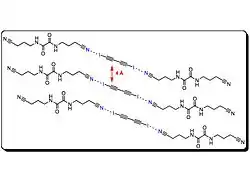
Structures poreuses
Les structures poreuses ont de nombreuses utilisations. De nombreux travaux se sont attelés à l'amélioration des réseaux métal-structure organique pour le stockage de l'hydrogène dans les voitures à hydrogène. Ces complexes d'inclusion à haut degré d'organisation ont des applications potentiels en catalyse et séparation moléculaire. L'organisation moléculaire est souvent contrôlée par l'utilisation de liaisons intermoléculaires comme la liaison hydrogène. Cependant, l'utilisation de ces liaisons hydrogène limite souvent la taille des pores accessibles pour des raisons d'empilement.
Pigge, et al., utilisent des liaisons halogènes entre des amines, des hétérocycles azotés, des carbonyles et d'autres halogénures organiques pour obtenir des structures poreuses. Ces réseaux cristallins organiques via des liaisons halogènes sont cependant assez rares car ce type de liaison est faible[23].
Les liaisons halogènes dans les systèmes biologiques
Notes et références
- P.A. Kollman et L.C. Allen, « Theory of the Hydrogen Bond », Chem. Rev., vol. 72, no 3, , p. 283-303 (lire en ligne)
- P. Metrangolo et G. Resnati, « Halogen Bonding: A Paradigm in Supramolecular Chemistry », Chem. Eur. J., vol. 7, no 12, , p. 2511 - 2519 (lire en ligne)
- (en) Ning Guo, Rémi Maurice, David Teze, Jérôme Graton, Julie Champion, Gilles Montavon et Nicolas Galland, « Experimental and computational evidence of halogen bonds involving astatine », Nature Chemistry, vol. 10, no 4, , p. 428–434 (ISSN 1755-4330 et 1755-4349, DOI 10.1038/s41557-018-0011-1, lire en ligne, consulté le )
- P. Metrangolo, H. Neukirch, T. Pilati et G. Resnati, « Halogen Bonding Based Recognition Processes: A World Parallel to Hydrogen Bonding », Acc. Chem. Res., vol. 38, no 5, , p. 386–395 (lire en ligne)
- (en) O. Hassel et Chr. Rømming, « Direct structural evidence for weak charge-transfer bonds in solids containing chemically saturated molecules », Q. Rev. Chem. Soc., vol. 16, no 1, , p. 1–18 (ISSN 0009-2681, DOI 10.1039/qr9621600001, lire en ligne, consulté le )
- F. Guthrie, « XXVIII. On the Iodide of Iodammonium », J. Chem. Soc., vol. 16, , p. 239–244 (lire en ligne)
- R. S. Mulliken, « Electronic Population Analysis on LCAO-MO Molecular Wave Functions. I », J. Chem. Phys., vol. 23, , p. 1833-1840. (lire en ligne)
- R. S. Mulliken, « Electronic Population Analysis on LCAO-MO Molecular Wave Functions. II. Overlap Populations, Bond Orders, and Covalent Bond Energies », J. Chem. Phys., vol. 23, , p. 1841-1846. (lire en ligne)
- O Hassel et J Hvoslef, « The Structure of Bromine 1,4-Dioxanate », Acta Chem. Scand., vol. 8, , p. 873-873 (lire en ligne)
- O Hassel, « Structural Aspects of Interatomic Charge-Transfer Bonding », Science, vol. 170, , p. 497–502 (lire en ligne)
- C. Møller et M. S. Plesset, « Note on an Approximation Treatment for Many-Electron Systems », Phys. Rev., vol. 46, no 7, , p. 618-622 (lire en ligne)
- P. Politzer, P. Lane, M. C. Concha, Y. Ma et J. S. Murray, « An overview of halogen bonding », J. Molecul. Model., vol. 13, no 2, , p. 305-311 (lire en ligne)
- (en) A. C. Legon, Halogen Bonding : Fundamentals and Applications, vol. 126, Berlin Heidelberg, P. Metrangolo & G.Resnati, coll. « Structure and Bonding », , 230 p. (ISBN 978-3-540-74329-3, LCCN 2007936673, lire en ligne), p. 17-64
- J. Xu, X. Liu, J. K.-P. Ng, T. Lin et C. He, « Trimeric supramolecular liquid crystals induced by halogen bonds », J. Mater. Chem., vol. 16, no 45, , p. 3540 - 3545 (lire en ligne)
- M. Weingarth, N. Raouafi, B. Jouvelet, L. Duma, G. Bodenhausen, K. Boujlel, B. Scöllhorn et P. Tekley, « Revealing molecular self-assembly and geometry of non-covalent halogen bonding by solid-state NMR spectroscopy », Chem. Commun., no 45, , p. 5981-5983 (lire en ligne)
- F. Meyer et P. Dubois, « Halogen bonding at work: recent applications in synthetic chemistry and materials science », Crystengcomm, vol. 15, , p. 3058–3071 (DOI 10.1039/C2CE26150B, lire en ligne)
- D. Braga, G. R. Desiraju, J. S. Miller, A. G. Orpen et S. L. Price, « Innovation in crystal engineering », CrysEngComm, vol. 4, , p. 500-509 (lire en ligne)
- G. M. J. Schmidt, « Photodimerization in the solid state », Pure Appl. Chem., vol. 27, no 4, , p. 647–678 (lire en ligne)
- P. Metrangolo, G. Resnati, T. Pilati, R. Liantonio et F. Meyer, « Engineering Functional Materials by Halogen Bonding », J. Polym. Sci. Part A: Polym. Chem., vol. 45, no 1, , p. 1–14 (DOI 10.1002/pola.21725, lire en ligne)
- H. Loc Nguyen, P. N. Horton, M. B. Hursthouse, A. C. Legon et D. W. Bruce, « Halogen Bonding: A New Interaction for Liquid Crystal Formation », J. Am. Chem. Soc., vol. 126, no 1, , p. 16–17 (DOI 10.1021/ja036994l, lire en ligne)
- D. W. Bruce, « The Materials Chemistry of Alkoxystilbazoles and Their Metal Complexes », Adv. Inorg. Chem, vol. 52, , p. 151–204 (DOI 10.1016/S0898-8838(05)52003-8)
- A. Sun, J. W. Lauher et N. S. Goroff, « Preparation of Poly(Diiododiacetylene), an Ordered Conjugated Polymer of Carbon and Iodine », Science, vol. 312, , p. 1030–1034 (DOI 10.1126/science.1124621)
- F. Pigge, V. Vangala, P. Kapadia, D. Swenson, N. Rath et Comm Chem, Hexagonal Crystalline Inclusion Complexes of 4-Iodophenoxy Trimesoate, vol. 38, , 4726–2728 p.
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Halogen bond » (voir la liste des auteurs).
 Portail de la chimie
Portail de la chimie