Hypercholestérolémie familiale
L’hypercholestérolémie familiale est une maladie génétique responsable d'une augmentation importante des taux sanguins de cholestérol (essentiellement celui du lipoprotéine de basse densité). Le risque principal est la survenue de maladies cardiovasculaires à un âge plus jeune que pour le reste de la population.
| Spécialité | Endocrinologie |
|---|
| CIM-10 | E78.0 |
|---|---|
| OMIM | 143890 |
| DiseasesDB | 4707 |
| MedlinePlus | 000392 |
| eMedicine | 121298 |
| MeSH | D006938 |
| Médicament | Statine, colésévélam (en), rosuvastatine et ézétimibe |
![]() Mise en garde médicale
Mise en garde médicale
Historique
Le rapport entre hypercholestérolémie importante et maladie cardiovasculaire est suspecté dès les années 1930[1], les formes familiales étant identifiées une décennie plus tard[2]. La transmission autosomique dominante a été mise en évidence en 1964[3] et la responsabilité d'une mutation sur le gène LDLR dans les années 1980[4]. Les travaux de Joseph Goldstein et de Michael Brown sur ce récepteur leur ont valu le prix Nobel de physiologie ou médecine en 1985.
Cause
Il s'agit d'une maladie génétique à transmission autosomique dominante. Plusieurs gènes sont concernés, codant des protéines intervenant dans la synthèse et l'élimination des LDL : récepteurs aux LDL (LDLR), apolipoprotéine B (APOB), et proprotéine convertase subtilisin/kexin de type 9 (PCSK9). Plus de 1000 mutations sur le gène LDLR ont été identifiées comme pouvant être responsable de l'hypercholestérolémie familiale[5].
Suivant le type de mutation, le niveau sanguin de cholestérol et sa traduction clinique ou évolutive peuvent être plus ou moins importants[6].
Épidémiologie
La forme hétérozygote concerne une personne sur 500[7] dans la population générale. Elle serait près du double aux États-Unis[8]. La forme homozygote concerne 1/160000-1/1000000 personnes.
Clinique
La principale conséquence de cette maladie est une élévation marquée des taux sanguins de LDL-Cholestérol (généralement 2-6 g/L dans la forme hétérozygote et 6-12 g/L dans la forme homozygote).
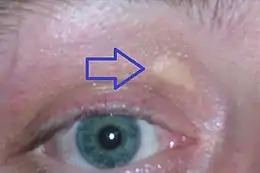
L'examen clinique est généralement pauvre. Il peut toutefois parfois constater la présence de xanthomes (petites tuméfactions jaunâtres sous-cutanée consistant, appelés xanthelasmas lorsqu'elles affectent les paupières), un arc cornéen, le tout témoignant de dépôts graisseux localisés. Ces xanthomes sont plus fréquemment retrouvés au dos des mains et au niveau des tendons d'Achille mais ces derniers apparaissent vers la troisième décennie de la vie[9].
Évolution
En l'absence d'un traitement adapté il existe dans cette maladie un risque très augmenté de survenue de maladies cardiovasculaires précoces. Dans la forme hétérozygote le risque d'être atteint d'une pathologie cardiovasculaire est d'environ 50% à 50 ans chez les hommes et à 60 ans chez les femmes.
L'atteinte de la forme homozygote est plus sévère et conduisait avant la mise au point de traitements efficaces à un décès par maladie coronarienne avant l'âge de 20-30 ans[10].
Traitement
Plusieurs recommandations ont été publiées sur le sujet. Celles de l'« European Atherosclerosis Society » datent de 2013[11]. Celles du « National Lipid Association » datent de 2011[12].
Ce sont essentiellement les statines. Ces dernières permettent de réduire le risque cardiovasculaire jusqu'à un niveau proche d'une population indemne de cette atteinte[13].
D'autres molécules sont en cours de test, comme le mipomersen[14] ou le lomitapide[15], permettant d'abaisser le LDL cholestérol, mais sans, pour l'instant, le recul pour démontrer une baisse du risque d'événements cardiovasculaires. Les anti-PCSK9 sont plus efficaces que les statines, dans la diminution du taux sanguin du cholestérol, certains ayant démontré une diminution du risque cardiovasculaire[16].
Évolocumab s'est révélé efficace pour faire baisser le taux de LDL cholestérol de 75 % [17]
Le dépistage d'un cas doit mener au dosage du cholestérol sanguin aux autres membres de la famille.
Notes et références
- Muller C, Xanthomata, hypercholesterolemia, angina pectoris, Acta Med Scand, 1938;89:75–84
- Wilkinson CF Jr, Hand EA, Fliegelman MT, Essential familial hypercholesterolemia, Ann Intern Med, 1948;29:671–686
- Khachadurian AK, The inheritance of essential familial hypercholesterolemia, Am J Med, 1964;37:402–407
- Brown MS, Goldstein JL, A receptor-mediated pathway for cholesterol homeostasis, Science, 1986;232:34–47
- Leigh SE, Foster AH, Whittall RA, Hubbart CS, Humphries SE, Update and analysis of the University College London low density lipoprotein receptor familial hypercholesterolemia database, Ann Hum Genet, 2008;72:485-498
- Gidding SS, Champagne MA, de Ferranti SD et al. The Agenda for familial hypercholesterolemia: A scientific statement from the american heart association, Circulation, 2015;132:2167-2192
- Marks D, Thorogood M, Neil HA, Humphries SE, A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia, Atherosclerosis, 2003;168:1-14
- de Ferranti SD, Rodday AM, Mendelson MM, Wong JB, Leslie LK, Sheldrick RC, Prevalence of familial hypercholesterolemia in the 1999 to 2012, United States National Health and Nutrition Examination Surveys (NHANES), Circulation, 2016;133:1067-1072
- Gill PJ, Harnden A, Familial hypercholesterolaemia, BMJ, 2012;344:e3228
- Scientific Steering Committee on behalf of the Simon Broome Register Group, Risk of fatal coronary heart disease in familial hypercholesterolaemia, BMJ, 1991;303:893-6
- Nordestgaard BG, Chapman MJ, Humphries SE et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society, Eur Heart J, 2013;34:3478–390a
- Goldberg AC, Hopkins PN, Toth PP et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia, J Clin Lipidol, 2011;5:133–140
- Versmissen J, Oosterveer DM, Yazdanpanah M et al. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study'', BMJ, 2008;337:a2423
- Raal FJ, Santos RD, Blom DJ et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial, Lancet, 2010;375:998-1006
- Cuchel M, Meagher EA, Theron HdT et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study, Lancet, 2013;381:40-46
- Shrank WH, Barlow JF, Brennan TA, « New Therapies in the Treatment of High Cholesterol », JAMA, 2015;314:1443-1444
- N Engl J Med 2020; 383:1317-1327 DOI: 10.1056/NEJMoa2019910 https://www.nejm.org/doi/full/10.1056/NEJMoa2019910?query=TOC.
 Portail de la médecine
Portail de la médecine