Dégénérescence lobaire frontotemporale
La dégénérescence lobaire frontotemporale (DLFT), dégénérescence fronto-temporale (DFT) ou démence fronto-temporale (DFT) constitue un ensemble de maladies neurologiques causées par l'atrophie dégénérative focale d'un ou plusieurs lobes cérébraux. Les manifestations de ces maladies dépendent des parties du cerveau concernées par l'atrophie. Ces manifestations sont très différentes de la maladie d'Alzheimer même si l'évolution peut se faire vers une démence. L'autre point commun est le phénomène dégénératif et l'absence de traitement efficace.
Pour les articles homonymes, voir DFT.
| Spécialité | Neurologie et endocrinologie |
|---|
| CISP-2 | N99 |
|---|---|
| CIM-10 | G23.0 |
| CIM-9 | 331.19 |
| OMIM | 600274 |
| DiseasesDB | 10034 |
| MeSH | D003704 |
![]() Mise en garde médicale
Mise en garde médicale
Le diagnostic se fait sur la clinique et l'imagerie médicale. Des marqueurs biologiques dans le liquide cérébrospinal seront de plus en plus utilisés dans un avenir proche.
L'examen anatomopathologique du cerveau est cependant le seul moyen de distinguer avec certitude les différents types de DLFT, car la corrélation demeure imprécise entre les symptômes, l'imagerie et les constatations neuropathologiques.
Les limites entre les différentes pathologies sont encore floues.
Ces maladies sont rarement héréditaires et quelques gènes commencent à être mis en évidence.
Historique
Le premier cas a été décrit en 1892 par Arnold Pick. Cette entité a été nommée par Alois Alzheimer « maladie de Pick » en 1911[1].
Épidémiologie
Il s'agit de la troisième cause de démences avant l'âge de 65 ans (après la maladie d'Alzheimer et les démences vasculaires[2]). Sa prévalence (avant l'âge de 65 ans) est comprise entre 1 et 26 cas pour 100 000 patients[1]. L'âge de survenue est essentiellement entre 45 et 65 ans mais un tiers des cas surviennent après 65 ans[1].
Diagnostic
Clinique/symptômes
Les signes cliniques de DLFT sont variables et permettent de définir les différents phénotypes rencontrés. Certains patients ne peuvent d'ailleurs pas être classés au début de la maladie. Les critères de diagnostic de cette maladie ont fait l'objet d'un consensus[3].
Avec ces éléments de diagnostic sont définis trois catégories.
Démence frontotemporale
Celle-ci se manifeste par une modification progressive du caractère et du comportement. Le plus souvent il s'agit d'une désinhibition. Le patient perd les « bonnes manières » en public. Il devient moins tolérant, plus rigide sur ses habitudes et les horaires. Des idées et des activités répétitives et des comportements d'amassage peuvent être également perçus. Elle est également appelée variant frontal ou variant comportemental de la DLFT.
Aphasie primaire progressive non fluente
Elle se manifeste par des difficultés de langage. Elle est dite non fluente car la vitesse de conversation ralentit. Les phrases sont moins longues. Le patient bute sur les mots. Il fait des erreurs de prononciation. La compréhension est au départ bien conservée. Certains patients présentent donc plus des difficultés d'articulation et des auteurs les isolent sous le terme d'anarthrie progressive. Un sous-type est également décrit comme apraxie de la parole selon des critères neuropsychologiques plus fins[4].
Démence sémantique
Il ne s'agit pas réellement d'une démence (en tout cas à la phase de début) et il est de plus en plus question de « dégénérescence sémantique »[5]. Elle se manifeste par une perte des concepts, les patients ne reconnaissant plus les objets qu'ils voient, qu'ils touchent, les mots qu'ils lisent ou qu'ils entendent. Ces objets semblent avoir disparu de leur connaissance. Parfois cela prend l'aspect d'un trouble de la mémoire ou du langage. Les patients gardent un certain temps la capacité de vivre seul d'où le terme inapproprié de démence. Elle est encore considérée par certains auteurs comme une forme d'aphasie primaire progressive (notamment aux États-Unis). Cette forme est alors dite fluente par opposition à l'aphasie progressive primaire non fluente.
Au-delà des critères
L'aphasie logopénique est une troisième forme d'aphasie primaire progressive (si l'on inclut dans ce groupe l'aphasie non fluente et la démence sémantique)[6]. Elle correspond en général en anatomopathologie à une maladie d'Alzheimer[7].
La dégénérescence cortico-basale et la paralysie supranucléaire progressive ont leurs symptômes propres mais peuvent débuter par des symptômes de démence fronto-temporale ou par ceux de l'aphasie primaire progressive. Elles peuvent à ce titre être incluses dans les DLFT.
Imagerie cérébrale
L'imagerie par résonance magnétique met en évidence une atrophie focale et d'allure lobaire : c'est ce qui fait la caractéristique radiologique de ces affections. La localisation de l'atrophie est en rapport avec les symptômes présentés par le patient, et inversement :
- dans la démence frontotemporale ou forme frontale de DLFT, ce sont les lobes frontaux qui sont les plus atteints[réf. souhaitée] ce qui explique les troubles du comportement. La partie antérieure des lobes temporaux est également touchée et participe à ces troubles ;
- dans l'aphasie primaire progressive, l'atrophie prédomine sur les régions du langage c'est-à-dire les régions fronto-temporales gauches[réf. souhaitée] ;
- dans la démence sémantique : l'atrophie est essentiellement temporale unilatérale[réf. souhaitée]. Le tableau clinique peut d'ailleurs être influencé par la latéralité de l'atteinte. Une atrophie frontale peut y être associée et ne manque pas d'apparaître au cours de l'évolution ;
- dans la paralysie supranucléaire progressive,
- dans la dégénérescence cortico-basale,
Cet examen permet d'éliminer une démence d'origine vasculaire ou une cause tumorale.
Des examens fonctionnels tels la scintigraphie cérébrale permettent aussi d'approcher le diagnostic, surtout si l'IRM ne montre pas d'anomalie significative. Ils montrent le fonctionnement des différentes parties du cerveau, par exemple en représentant la quantité de glucose consommée ou le débit sanguin. D'une façon générale, ce débit est moindre dans les régions concernées par la maladie : le lobe frontal pour la démence fronto-temporale, le lobe temporal pour la démence sémantique, la région périsylvienne pour une aphasie progressive. Des chevauchements sont toutefois possibles et c'est surtout le lieu de la maladie qui influence le résultat plus que son type.
Tests neuropsychologiques
Ces tests mettent surtout en évidence l'absence d'amnésie importante, l'absence de désorientation spatio-temporelle, ainsi que l'altération des fonctions exécutives, signe de l'atteinte frontale du patient. Ils permettent également d'évaluer le langage, afin de détecter une démence sémantique ou une aphasie. Ils précisent les déficits spécifiques à chaque phénotype.
Biologie
Les examens biologiques sanguins et du liquide céphalorachidien éliminent une cause inflammatoire ou métabolique. Des avancées sont à prévoir grâce au dosage de la protéine tau (phosphorylée) et de la substance bêta amyloïde dans le LCR qui pourraient permettre de différencier les DLFT de la maladie d'Alzheimer. Ils sont par contre incapables de différencier les différents sous-types de DLFT.
Examen neuropathologique post-mortem/Anatomopathologie
Comme pour les autres maladies neurodégénératives, il reste, à ce jour, le seul examen permettant de porter un diagnostic définitif de démence frontotemporale et de connaître son sous-type. À cet égard, les patients et leur famille qui font don de leur cerveau[8] permettent des avancées considérables pour connaître et lutter contre les DFLT et les maladies neurodégénératives.
Différents types anatomopathologiques

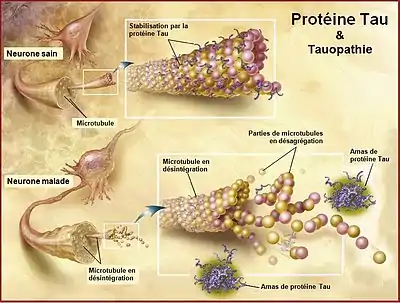
L'anatomopathologie des DLFT à beaucoup évolué ces dernières années. Cette évolution se fait conjointement aux découvertes dans le domaine génétique[9].
Toutes ces avancées tendent à remanier régulièrement les critères et les classifications de ces maladies ce qui ne facilite pas toujours la clarté. À titre d'exemple la dégénérescence cortico-basale a une définition clinique et une définition anathomopathologique. Un patient souffrant de dégénérescence cortico-basale (clinique) n'aura pas forcément une atteinte cérébrale de type dégénérescence cortico-basale (anatomopathologique). Reste à savoir si, dans le but d'un traitement, c'est la clinique, l'imagerie, l'anatomopathologie ou la génétique qui définissent le mieux ou de la façon la plus pertinente et efficiente la maladie du patient[10].
Les deux types anatomopathologiques les plus répandus (environ 90 % des cas) sont les pathologies de la protéine Tau et les pathologies de la protéine TDP-43 (TAR DNA binding Protein 43). Chacune peut être divisée en sous types distincts. Par exemple, quatre sous types de DLFT avec des inclusions tau sont connus. Ils se distinguent entre eux par les propriétés biochimiques de l'inclusion tau (4R ou 3R), par la distribution et l'aspect des inclusions ou par les modifications cellulaires engendrées. Ils incluent : paralysie supranucléaire progressive, dégénérescence cortico-basale, maladie de Pick et démence frontotemporale liée au chromosome 17.
Les 10 % restants représentent un groupe hétérogène mais ils semblent avoir en commun une pathologie de la protéine FUS (FUsed in Sarcoma)[11]. Ces derniers cas regroupent pour une bonne part les cas de pathologie de l'ubiquitine qui définissaient encore, il y a peu, ces 10 % mais dont l'avancée des recherches tend à réduire le nombre.
Génétique
Près de 40 % des patients souffrant de DLFT ont une histoire familiale, mais un quart d'entre eux sont évocateurs d'une transmission autosomique dominante[12]. Plusieurs gènes sont reconnus associés à ces pathologies[13].
Les trois gènes les plus représentés sont le gène C9orf72 (retrouvé dans la majorité des DFLT associées à la SLA avec une pathologie TDP-43) , le gène de la progranuline, GRN, (associé à une pathologie TDP-43), le gène MAPT (pathologie Tau). À eux trois ils expliquent plus de la moitié des DFLT familiales[14]. Les gènes CHMP2B, VCP, TARDBP, FUS sont beaucoup moins représentés. Malgré cela ces maladies ne sont pas héréditaires dans la majorité des cas.
Traitement
Il n'y a malheureusement pas de traitement curatif de ces pathologies. Aucun moyen actuel ne permet d'empêcher l'accumulation des protéines pathologiques (ou plutôt dont l'accumulation est pathologique).
Les médicaments utilisés dans la maladie d'Alzheimer ne sont pas efficaces. Il s'agit donc d'un traitement symptomatique au cas par cas en essayant de ne pas faire subir d'effets secondaires au patient (voir Dégénérescence cortico-basale et Paralysie supranucléaire progressive pour le détail sur ces pathologies). Les traitements psychotropes augmentant la sérotonine sont recommandés, en particulier la trazodone, délivrée en France par les pharmacies des hôpitaux, selon la procédure des autorisations transitoires d'utilisation (ATU).[réf. souhaitée][15],[16]
Les prises en charge non médicamenteuses (kinésithérapie, ergothérapie, orthophonie, neuropsychologie, accompagnement psychologique et matériel de l'aidant) sont souvent les plus importantes.
Pronostic
La durée de survie après l'apparition des premiers symptômes est comprise entre 6 et 11 ans, les formes avec atteintes motrices étant les plus graves[1].
Organisation des soins

En France, depuis juillet 2007, dans le cadre du Plan national Maladies rares[17] (PNMR), un centre de référence et son réseau national de centres de compétence ont été labellisés pour la prise en charge des personnes atteintes de démence fronto-temporale.
Le réseau des centres de référence et de compétence pour les DFT[18], la coordination est faite à l'hôpital de la Salpêtrière à Paris, il travaille avec ses 12 Centres de Compétence régionaux, pour améliorer la prise en charge des patients et des familles sur toute la France.
Depuis le , le PNMR (plan national Maladies rares)[19] a été reconduit.
Malades célèbres
Certains auteurs suggèrent que le philosophe Friedrich Nietzsche aurait souffert de dégénérescence lobaire frontotemporale (DFLT) plutôt que de neurosyphilis[20].
Le réalisateur Curtis Hanson était atteint de DFLT, qui l'a emporté en 2016[21] tout comme l'actrice Charmian Carr[22].
Notes et références
- Bang J, Spina S, Miller BL, Frontotemporal dementia, Lancet, 2015;386:1672–1682
- Vieira RT, Caixeta L, Machado S et al. Epidemiology of early-onset dementia: a review of the literature, Clin Pract Epidemiol Ment Health, 2013;9:88–95
- (en) Mc Kahnn GM, Albert MS, Grossman M, Miller BL, Dickson D, Trojanowski JQ. « Clinical and pathological diagnosis of frontotemporal dementia. Report of the Work Group on Frontotemporal Dementia and Pick’s Disease » Archives of Neurology 2001;58:1803-9. .
- (en) Rohrer JD, Knight WD, Warren JE, Fox NC, Rossor MN, Warren JD, « Word-finding difficulty: a clinical analysis of the progressive aphasias » Review Brain. 2008 Jan;131(Pt 1):8-38. .
- Moreaud O, Belliard S, Snowden J, Auriacombe S, Virat-Brassaud ME et al., « Démence sémantique : réflexions d’un groupe de travail pour des critères de diagnostic en français et la constitution d’une cohorte de patients [Semantic dementia: reflexions of a French working group for diagnostic criteria and constitution of a patient cohort] », Rev Neurol (Paris), vol. 164, no 4, , p. 343-53. (PMID 18439926, DOI 10.1016/j.neurol.2008.02.031, résumé).
- (en) Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, Ogar JM, Rohrer JD, Black S, Boeve BF, Manes F, Dronkers NF, Vandenberghe R, Rascovsky K, Patterson K, Miller BL, Knopman DS, Hodges JR, Mesulam MM, Grossman M, « Classification of primary progressive aphasia and its variants », Neurology, vol. 76, no 11, , p. 1006-14. (PMID 21325651, PMCID PMC3059138, DOI 10.1212/WNL.0b013e31821103e6, lire en ligne [html]).
- (en) Mesulam M, Wicklund A, Johnson N, Rogalski E, Léger GC, Rademaker A, Weintraub S, Bigio EH, « Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia », Ann Neurol, vol. 63, no 6, , p. 709-19. (PMID 18412267, PMCID PMC2858311, DOI 10.1002/ana.21388, lire en ligne [html]).
- Site d'information sur le don de cerveau.
- (en) Mackenzie I.R. et al. « Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update » Acta Neuropathologica 2010;119:1-4. .
- (en) Rohrer JD et al. « Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration » Brain 2011;134:2565-81. .
- (en) Mackenzie IR, Munoz DG, Neumann M. et al. « Distinct pathological subtypes of FTLD-FUS » Acta Neuropathol. 2011;121(2):207-18. .
- Rohrer JD, Guerreiro R, Vandrovcova J et al. The heritability and genetics of frontotemporal lobar degeneration, Neurology, 2009;73:1451–1456.
- (en) Sieben A. et al. « The genetics and neuropathology of frontotemporal lobar degeneration » Acta Neuropathol. 2012;124:353-72. .
- Le Ber I, Genetics of frontotemporal lobar degeneration: an up-date and diagnosis algorithm, Rev. Neurol. (Paris), 2013;169:811–819.
- Florence Lebert, Willy Stekke, Christine Hasenbroekx et Florence Pasquier, « Frontotemporal Dementia: A Randomised, Controlled Trial with Trazodone », Dementia and Geriatric Cognitive Disorders, vol. 17, no 4, , p. 355–359 (ISSN 1420-8008 et 1421-9824, DOI 10.1159/000077171, lire en ligne, consulté le )
- Jee Bang, Salvatore Spina et Bruce L Miller, « Frontotemporal dementia », The Lancet, vol. 386, no 10004, , p. 1672–1682 (ISSN 0140-6736, DOI 10.1016/s0140-6736(15)00461-4, lire en ligne, consulté le )
- [PDF] Plan National Maladies Rares 2005-2008.
- Réseau des Centres de Référence et de Compétence pour les DFT.
- Plan National Maladies Rares 2010-2014.
- (en) Orth M, Trimble MR, « Friedrich Nietzsche's mental illness--general paralysis of the insane vs. frontotemporal dementia », Acta Psychiatr Scand, vol. 114, no 6, , p. 439-44; discussion 445. (PMID 17087793, DOI 10.1111/j.1600-0447.2006.00827.x)
- (en) « Curtis Hanson, Director of ‘L.A. Confidential,’ Dies at 71 », sur variety.com, 20 septembre 2016
- (en-US) « Charmian Carr, Liesl von Trapp in ‘The Sound of Music’ Film, Dies at 73 (Published 2016) », The New York Times, (ISSN 0362-4331, lire en ligne, consulté le )
Voir aussi
Bibliographie
- (en) Manuela Neumann, Frontotemporal dementia in Orphanet [PDF]
- (en) John C Van Swieten, Peter Heutink, « Frontotemporal Dementia with Parkinsonism » In Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993-2005
Liens externes
 Portail de la médecine
Portail de la médecine