Communication interventriculaire
La communication interventriculaire (en abrégé CIV) est la malformation cardiaque congénitale la plus fréquente après la CIA : elle représenterait près de 10 % de l'ensemble des cardiopathies congénitales chez l'humain.
Pour les articles homonymes, voir CIV.
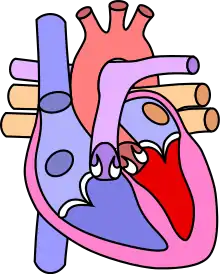

Dans le cœur normal, la cloison qui sépare les ventricules droit et gauche, dénommée « septum interventriculaire », est totalement hermétique. Une CIV correspond à la présence d'un orifice plus ou moins large dans cette cloison, permettant le passage direct du sang d'un ventricule à l'autre. Les conséquences de cette malformation dépendent essentiellement de la taille de la communication. Dans la plupart des cas, l'orifice est petit : ces CIV sans conséquence réellement gênante sont appelées « maladie de Roger ». L'évolution d'une CIV dépend de sa localisation sur le septum, certaines étant susceptibles de se fermer spontanément dans les mois ou années suivant la naissance.
Cet article ne s'intéresse qu'aux CIV congénitales isolées, les plus fréquentes. Il ne traitera pas des CIV qui ne sont qu'un des éléments constitutifs de malformations cardiaques plus complexes telles que la tétralogie de Fallot, ni des CIV acquises, c'est-à-dire constituées secondairement et qui viennent compliquer l'évolution d'un infarctus du myocarde ou d'une endocardite infectieuse.
Conséquences
Avant la naissance
La présence d'une CIV isolée n'a habituellement pas de conséquence avant la naissance. La circulation fœtale repose en effet déjà sur la présence de deux communications entre le cœur droit et le cœur gauche, le foramen ovale à l'entrée du cœur, le canal artériel à sa sortie. À condition qu'il n'y ait pas d'autre malformation présente, l'existence d'une communication supplémentaire, située entre les deux précédentes, ne modifie que très peu la circulation du sang et ceci d'autant plus que chez le fœtus, la pression sanguine est pratiquement équivalente dans les deux ventricules. La quantité de sang qui passe par la CIV est donc minime, habituellement dirigée du ventricule droit vers le ventricule gauche, en particulier au troisième trimestre de la grossesse.
Après la naissance
Normalement, le foramen ovale et le canal artériel se ferment et il s'établit progressivement une différence de pressions entre le cœur droit (oreillette droite, ventricule droit et artères pulmonaires) et le cœur gauche (oreillette gauche, ventricule gauche et aorte). La pression tend à diminuer fortement dans le cœur droit, en particulier le ventricule droit (où la pression passe de 60 mmHg environ à 20–25 mmHg environ). Elle reste stable puis augmente dans le cœur gauche et en particulier dans le ventricule gauche où son évolution peut être appréciée de façon simple par la mesure de la tension artérielle. Le passage de sang au travers de la CIV tend donc à augmenter, son importance étant fonction de la taille de l'orifice et de la différence de pression de part et d'autre de la communication.
Notion de shunt gauche-droit

On parle de shunt — « court-circuit » — quand une partie du sang emprunte un « chemin » plus court que celui qu'il aurait dû parcourir normalement. L'expression « gauche-droit » caractérise le sens du shunt, dans le cas présent, du ventricule gauche vers le ventricule droit.
Par sa fréquence, la CIV est le modèle des cardiopathies congénitales responsables d'un shunt gauche-droit, mais elle n'est pas la seule. En fait, toute communication isolée entre le cœur gauche et le cœur droit sera responsable d'un shunt gauche-droit. Les plus fréquentes sont :
- les communications interauriculaires entre les deux oreillettes ;
- les communications interventriculaires entre les deux ventricules ;
- la persistance du canal artériel entre l'aorte et l'artère pulmonaire.
D'autres malformations cardiaques, plus rares, sont également responsables d'un shunt gauche-droit :
- canal atrio-ventriculaire (dans une forme partielle ou complète) ;
- fistule ou fenêtre aorto-pulmonaire ;
- communication ventricule gauche-oreillette droite ;
- fistules coronaro-cardiaques ou coronaro-pulmonaires.
Surcharge de la vascularisation pulmonaire et du cœur gauche
En présence d'une CIV, le sang présent dans le ventricule gauche peut emprunter le parcours normal, vers l'aorte et l'ensemble du corps (« grande » circulation ou circulation « systémique »), ou passer à travers la CIV vers le ventricule droit pour retourner directement vers les poumons d'où il vient (« petite » circulation ou circulation « pulmonaire »).
La quantité de sang qui passe par la CIV et emprunte le court-circuit est d'autant plus importante que la CIV est large et que la différence de pression entre les deux ventricules est importante. On parle de « CIV restrictive » quand la taille de l'orifice est suffisamment réduite pour s'opposer par elle-même au passage d'une grande quantité de sang. En pratique, chez un nouveau-né, c'est le cas si le diamètre de la CIV est inférieur à 3 mm environ. Quand la CIV est plus large, elle n'oppose pratiquement aucune restriction au passage du sang (« CIV non restrictive ») et la quantité qui passera sera uniquement fonction de la différence de pression de part et d'autre de la communication.
Normalement, après la naissance, la baisse de la pression pulmonaire se fait de façon exponentielle, rapide dans les premières heures de vie, puis plus lente jusqu'à l'âge d'un mois environ. La différence de pression de part et d'autre de la CIV suit donc la même évolution et n'atteindra son niveau définitif que vers l'âge d'un mois (et parfois plus tard, en particulier en cas de shunt gauche-droit). Cela explique que la plupart des CIV n'aient que peu de retentissement dans les premiers jours de vie et qu'une éventuelle gêne fonctionnelle du nourrisson n'apparaisse que plus tard.
Cette gêne fonctionnelle sera liée à l'augmentation de la quantité de sang qui revient aux poumons puis, de là, dans les cavités gauches. Il faut savoir qu'en présence d'une CIV large, il peut passer 2, 3 voire 4 fois plus de sang par le court-circuit que par le chemin normal vers l'aorte. Cela veut dire que les poumons reçoivent 3, 4 ou 5 fois plus de sang que la normale (1 quantité normale + la quantité passant par le shunt). De même, à la sortie des poumons, l'oreillette puis le ventricule gauche doivent faire circuler jusqu'à 5 fois plus de sang que normalement : un nourrisson porteur d'une large CIV se trouve donc en permanence dans un état proche d'un effort maximal. Cet excès de sang au niveau des poumons peut provoquer un œdème pulmonaire et une insuffisance ventriculaire gauche.
Altérations de la vascularisation pulmonaire
Normalement, les artères pulmonaires ont une structure adaptée à un fonctionnement sous un faible régime de pression. En présence d'une CIV, elles doivent s'adapter à l'augmentation de débit. Elles le font d'abord par une vasodilatation puis, si cela ne suffit, par une augmentation de pression. Dans les premiers mois de vie, cette adaptation est réversible. La fermeture de la CIV s'accompagne d'un retour à la normale des artères pulmonaires (diamètre et pression). Si cette situation perdure, il se produit des modifications de la structure de ces vaisseaux qui les transforment de façon irréversible en artères faites pour fonctionner sous fortes pressions. L'hypertension artérielle pulmonaire (HTAP) « dynamique » et réversible du début s'est transformée en une hypertension artérielle pulmonaire «fixée» et définitive. On parle alors de syndrome d'Eisenmenger, complication grave contre laquelle nous ne disposons que de peu de possibilités thérapeutiques.
Classiquement, une HTAP ne devient fixée qu'au-delà de l'âge de un an. En pratique cependant, cette évolution peut survenir plus tôt, en particulier chez les enfants porteurs d'une trisomie 21 (syndrome de Down). Exceptionnellement, elle peut s'installer dès la naissance par « persistance de résistances pulmonaires de type fœtal ».
Épidémiologie et causes
40 % des malformations cardiaques congénitales comportent une CIV[2]. Un dépistage systématique montre une incidence, à la naissance, atteignant 5 %, les neuf-dixièmes disparaissant spontanément après quelques semaines[3].
Les causes sont probablement une combinaison de facteurs environnementaux et de facteurs génétiques. Parmi les facteurs environnementaux suspects, on peut citer le diabète gestationnel, la phénylcétonurie[4].
Différentes localisations des CIV
La complexité de la formation embryologique de la cloison interventriculaire, nécessitant l'alignement et la fusion de quatre constituants explique, au moins en partie, la fréquence des CIV.
Connaître non seulement la taille mais aussi la localisation d'une CIV a un double intérêt :
- certaines localisations sont parfois associées à certaines anomalies génétiques et peuvent donc inciter à pratiquer une amniocentèse pour étude du caryotype fœtal si elles sont dépistées lors des échographies anténatales.
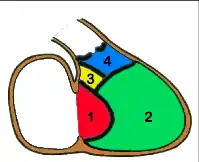
- C'est le cas des CIV qui siègent dans le septum d'admission (partie 1 en rouge sur le schéma), fréquemment associée à une Trisomie 21, et de celles qui siègent dans le septum sous-aortique (partie 4 en bleu sur le schéma), parfois associées à une anomalie du chromosome 22 (Microdélétion 22q11) ;
- dans certaines localisations, la CIV est susceptible de diminuer de taille, voire de se fermer totalement de façon spontanée alors que dans d'autres, ce mécanisme de guérison spontané n'est jamais observé.
- les CIV siégeant dans le septum membraneux (partie 3 en jaune sur le schéma) et encore plus les CIV musculaires situées dans la partie 2 (en vert sur le schéma) sont fréquemment capables de « se guérir toutes seules » (à condition que l'état de l'enfant permette d'attendre).
- les CIV situées dans les parties 1 (septum d'admission) et 4 (septum sous-aortique) ne se ferment jamais spontanément et devront donc, sauf exception, être opérées tôt ou tard.
.png.webp)
Schéma sur les localisations des communications interventriculaires (légende) :
- CIV membraneuse et péri-membraneuse (1-2-3)
- CIV d'admission ou postérieure (4)
- CIV musculaire ou trabéculée (5-6)
- CIV sous-aortique (7)
- CIV infundibulaire (8).
Anecdote : les CIV « infundibulaires », c'est-à-dire siégeant sous les valves pulmonaires sont fréquentes en Asie alors qu'elles sont très rares en Europe où elles pourraient être un lointain souvenir des invasions barbares. Elles ne se ferment jamais spontanément.
Diagnostic
Anténatal
Le diagnostic de CIV est possible pendant la grossesse, mais reste encore relativement rarement posé quand la communication est petite et de siège membraneux ou musculaire, ce qui n'est pas très grave puisque ces CIV n'ont pas de conséquence sur la grossesse ou l'accouchement et ne sont pas particulièrement associées à une anomalie chromosomique. Certains échographistes ne les recherchent d'ailleurs pas, et ce volontairement, estimant que ce diagnostic est une source inutile d'inquiétude parentale, d'autant qu'ils régressent dans près de la moitié des cas[5].
Aspects échographiques avant la naissance

Gestion de la grossesse
En soi, la découverte d'une CIV lors d'une échographie fœtale ne modifie ni le cours de la grossesse, ni les modalités d'accouchement et ne s'accompagne habituellement d'aucun retentissement sur la croissance du fœtus. Elle implique par contre :
- de préciser la localisation de cette CIV puisque certaines justifieront la réalisation d'une amniocentèse à la recherche d'une anomalie chromosomique chez le fœtus (voir ci-dessous) ;
- de tenter d'apprécier sa taille afin de définir les modalités de surveillance après la naissance et, si la CIV apparaît large, d'informer les parents sur la démarche thérapeutique dans les premiers mois de vie ;
- de rechercher d'éventuelles anomalies associées, soit cardiaques et susceptibles de modifier la symptomatologie, soit extra-cardiaques dans la crainte d'un syndrome polymalformatif.
CIV et amniocentèse
La décision finale de réaliser ou non une amniocentèse relève des parents et d'eux seuls : les médecins proposent, les parents disposent. Faire cet examen peut être considéré comme inutilement dangereux (même si les risques sont faibles, inférieurs à 1 %) si d'emblée les parents savent qu'ils ne pourraient se résoudre à demander une interruption médicale de grossesse en cas d'anomalie chromosomique. Le seul intérêt serait alors de permettre à la famille de se préparer à accueillir un enfant « anormal » (par exemple porteur d'une Trisomie 21) plutôt que d'affronter brutalement cette réalité dans les minutes qui suivent l'accouchement.
L'attitude médicale de proposer ou non une amniocentèse après la découverte d'une CIV ne fait pas encore l'objet d'un consensus parfait :
- tous s'entendent pour proposer cet examen en cas de :
- CIV isolée si elle est postérieure (risque de Trisomie 21) ou sous-aortique (risque de délétion 22q11),
- CIV associée à une ou plusieurs autre(s) anomalie(s), cardiaques ou extra-cardiaques,
- la plupart sursoit à cet examen si la CIV siège dans le septum musculaire, est unique, de petite taille et isolée ;
- l'attitude vis-à-vis des CIV du septum membraneux reste mal codifiée : en effet, ces CIV sont parmi les plus fréquentes et en règle isolées, mais certains enfants trisomiques peuvent ne présenter que ce « signe d'appel » lors des échographies anténatales et quelques CIV membraneuses pourraient s'accompagner d'une délétion 22q11.
CIV et interruption médicale de grossesse
Une CIV isolée n'amène pas à discuter d'une interruption médicale de grossesse (IMG), quelle que soit sa taille ou sa localisation, car il s'agit d'une malformation parfaitement curable. Une IMG ne pourrait être envisagée qu'en fonction d'autres anomalies associées, en particulier génétiques.
Cette attitude est d'autant plus fondée qu'il faut savoir qu'une CIV peut se fermer ou au moins se réduire spontanément pendant la grossesse elle-même. Ainsi, « il est urgent d'attendre » lorsqu'une CIV, même volumineuse, est découverte lors d'une échographie précoce, entre 12 et 18 semaines d'aménorrhée. Les bonnes surprises ne sont pas rares.
Prise en charge du nouveau-né
En l'absence de diagnostic anténatal, c'est habituellement la perception d'un souffle cardiaque à l'auscultation qui révèle l'existence d'une CIV.
Ce souffle est souvent absent lors des tout premiers jours de vie, en particulier quand le pédiatre examine le nouveau-né en salle de travail ou le lendemain de l'accouchement. Ceci est lié au fait que les pressions pulmonaires sont encore élevées et qu'il n'y a pas de shunt significatif au travers de la communication.
Le diagnostic de CIV est donc souvent retardé sans qu'il y ait eu d'erreur ou insuffisance médicale.
Quand le souffle est découvert, il est habituellement de faible intensité et reste isolé chez un enfant parfaitement bien portant par ailleurs, et ceci quelle que soit la taille de la communication. La réelle gravité ne pourra donc être appréciée que par la réalisation d'une échocardiographie, précisant la taille et la localisation de la communication.
En fonction des résultats de l'échocardiographie, on pourra distinguer :
- les petites CIV, habituellement musculaires (ou « trabéculées ») qui n'occasionneront aucun problème et ont toutes les chances de se guérir toutes seules en quelques semaines ou quelques mois. La surveillance clinique habituelle du nourrisson sera suffisante. C'est de loin le cas le plus fréquent ;
- les CIV plus larges, susceptibles d’entraîner une insuffisance cardiaque du nourrisson dans les quelques jours ou semaines suivant la sortie de maternité. Elles justifient une surveillance plus étroite, souvent un traitement médical (qui peut être assuré à domicile) et parfois un traitement chirurgical (habituellement après quelques mois). Cette éventualité est relativement rare et ne représente qu'environ 10 % de l'ensemble des CIV.
Clinique
La présentation clinique dépend de la taille et de la position de la CIV. Elle est donc très variable, allant du nourrisson parfaitement asymptomatique en dehors d'un souffle cardiaque isolé au tableau de grande insuffisance cardiaque.
L'auscultation cardiaque note habituellement un souffle systolique diffus (« en rayons de roue ») dont l’intensité est inversement proportionnelle à la taille de la communication. L'intensité du souffle ne reflète donc absolument pas la gravité de la CIV (on pourrait même écrire au contraire...). C'est en prêtant attention aux bruits qui entourent ce souffle que le cardiologue pourra se faire une idée de l'importance de la CIV :
- bruit de « galop » traduisant une augmentation de débit au travers de la valve mitrale ;
- éclat du deuxième bruit au foyer pulmonaire traduisant une élévation des pressions pulmonaires.
Dans une certaine mesure, les caractéristiques du souffle peuvent orienter vers le type anatomique de la CIV responsable :
- un souffle rude, intense, holosystolique (« durant toute la systole »), perçu au maximum au 4e espace intercostal gauche est très évocateur de CIV membraneuse ;
- un souffle de même nature mais plus haut situé dans le thorax fera rechercher une CIV infundibulaire ;
- un souffle plus doux, protomésosystolique (c'est-à-dire « se produisant au début et au milieu de la systole »), perçu à l'apex ou à l'endapex est évocateur de CIV musculaire. À l'inverse des deux précédents, le souffle d'une CIV musculaire peut facilement être méconnu chez un nourrisson agité.
Le nourrisson présente parfois des signes d'insuffisance cardiaque. se traduisant par des sueurs, en particulier sur le front et le cuir chevelu, très inhabituelles chez un nourrisson (cela peut aller d'un front un peu moite à des cheveux trempés comme à la sortie du bain) et une respiration rapide (polypnée) lors d’efforts tels que les pleurs ou la prise du biberon. Celle-ci est lente, entrecoupée de pauses et laisse des restes plus ou moins importants à chaque tétée. Ces difficultés d'alimentation s'accompagnent rapidement de troubles de la croissance.
La cyanose (coloration bleutée de la peau et des muqueuses) est normalement absente dans la CIV. Si une cyanose apparait, d'abord lors des colères puis de façon plus ou moins permanente, cela signifie :
- soit que le diagnostic initial de CIV est faux et qu'il s'agissait d'une Tétralogie de Fallot. C'est une éventualité non exceptionnelle si le diagnostic de CIV évoqué à la naissance sur l'auscultation n'a pas été vérifié et confirmé par une échocardiographie ;
- soit que la CIV se complique d'une hypertension pulmonaire fixée et qu'un syndrome d'Eisenmenger est en train de s'installer (voir plus haut).
Examens complémentaires

Trois examens peuvent être utiles, la radiographie du thorax, l'électrocardiogramme et l'échocardiographie.
L'échocardiographie-doppler
En pratique, l'échocardiographie doppler, examen non douloureux et non irradiant suffit pour poser un diagnostic précis de la malformation et sauf exception pour apprécier son retentissement et sa gravité. L'image ci-contre nous montre une turbulence sanguine (en vert) traversant la paroi inter ventriculaire (ligne verticale en gris), et allant d'un ventricule à l'autre (cavités ventriculaires en noir). Le doppler permet de quantifier le shunt (calcul du rapport du débit systémique sur débit pulmonaire). Cet examen permet également de détecter des anomalies cardiaques associées, d'évaluer le retentissement sur le ventricule droit et d'estimer les pressions des cavités cardiaques droites.
La radiographie du thorax
Obtenir un bon cliché (c'est-à-dire en inspiration forcée) chez le nouveau-né ou le nourrisson est une affaire délicate et tous les signes décrits ci-dessous peuvent être faussement présents sur un mauvais cliché.
- augmentation de l'ombre cardiaque avec débord de l'arc inférieur gauche (correspondant au ventricule gauche). Cette « cardiomégalie » est grossièrement proportionnelle à l'importance du débit à travers la communication.
- augmentation des opacités vasculaires pulmonaires, traduisant l'augmentation de débit dans les vaisseaux (artères, capillaires et veines) des poumons.
- parfois, aspects d'atélectasie ou de distension alvéolaire localisés, traduisant des troubles des échanges gazeux liés à la compression des voies aériennes (bronches et bronchioles) par les artères pulmonaires dilatées.
Électrocardiographie (ECG)
Cet examen reste normal dans les petites CIV. Lorsque le shunt est plus important, il peut montrer des signes d'hypertrophie (augmentation de la masse musculaire) du ventricule gauche, habituellement déjà bien objectivés par l'échocardiographie. L'ECG s'avère en fait utile essentiellement lorsque l'on veut s'assurer de l'absence d'hypertrophie du ventricule droit, moins bien analysée par l'échocardiographie et dont la présence témoignerait d'une élévation des pressions pulmonaires.
N.B : L'analyse de l'ECG pédiatrique diffère fortement de celle faite chez l'adulte. À ce titre, et sauf cas particulier, il ne faut pas tenir compte de l'interprétation automatique proposée par la plupart des appareillages actuels, source d'erreurs grossières.
Le cathétérisme cardiaque n'est plus guère fait, sauf avant une procédure de fermeture de la CIV par voie endo vasculaire.
L'IRM cardiaque est un examen intéressant chez l'adulte[6], permettant de visualiser le défect et de quantifier le shunt.
Classification des CIV
De nombreuses classifications des CIV ont été proposées, essayant de traduire la diversité de leurs manifestations cliniques et de leurs conséquences. Les plus anciennes, bien que restant imparfaites sont celles proposées par Nadas[7] et Nouaille[8] dont s'inspire fortement le tableau ci-dessous. La plus actuelle est celle de l'« International Society for Nomenclature of Paediatric and Congenital Heart Disease » datant de 2018[9].
N.B : Les CIV type IV, non abordées dans cet article, correspondent à des CIV de taille variable associées à un rétrécissement situé entre la CIV et les artères pulmonaires (sténose médioventriculaire du VD, sténose infundibulaire, sténose valvulaire pulmonaire). Cet obstacle, en élevant les pressions dans le ventricule droit, diminue voire annule le shunt gauche-droit et « protège » la vascularisation pulmonaire comme le ferait un cerclage chirurgical de l'artère pulmonaire (voir plus bas).
| symptôme | type Ia | type Ib | type IIa | type IIb | type III | type IV |
|---|---|---|---|---|---|---|
| dyspnée | absente | aux efforts intenses | permanente | aux efforts | variable | variable |
| insuffisance cardiaque | absente | absente | présente ++ | présente + | absente | absente |
| cyanose | absente | absente | absente | absente | présente | variable |
| souffle systolique | intense + | intense + | intense | modéré | faible/absent | lié à l'obstacle |
| rythme de galop | absent | absent | présent | absent/faible | absent | absent |
| éclat du B2 pulmonaire | absent | absent | faible/modéré | modéré/fort | intense | absent |
| vascularisation pulm. (Rx) | normale | faiblement augmentée | très augmentée | augmentée | diminuée, anormale | variable |
| cardiomégalie (Rx) | non | faible | importante | moyenne | non | non (variable) |
| taille de l'OG (écho) | normale | dilatation modérée | dilatation importante | dilatation importante | peu ou pas dilatée | peu ou pas dilatée |
| diamètre du VG (écho) | normal | dilatation modérée | dilatation importante | dilatée, variable | normal | normal |
| diamètre du VD (écho) | normal | normal | normal | normal | souvent augmenté | variable |
| shunt Gauche-Droit | minime | moyen | important | moyen/important | minime/absent | minime/absent |
| shunt droit-gauche | non | non | non | non | présent | variable |
| pressions pulmonaires. | normales | faiblement augmentées | augmentées | très augmentées | très augmentées | normales/basses |
| résistances pulmonaires. | normales | normales | peu augmentées | augmentées | très augmentées | normales |
| indication chirurgicale | non | rarement | oui | oui, urgente | non | variable |
Abréviations : B2 : deuxième bruit du cœur - OG : oreillette gauche - VG : ventricule gauche - VD : ventricule droit.
Traitement
Il est très variable, dépendant de la localisation, de la taille et de la tolérance clinique de la communication ainsi que du niveau des résistances pulmonaires. Le seul traitement commun à toutes les CIV, quelles qu'elles soient, est le traitement préventif des infections :
- vaccination anti-grippale à tout âge et vaccination contre les infections à VRS (virus respiratoire scyncitial) chez le nourrisson ;
- éradication de tout foyer infectieux, en particulier ORL ou dentaire, porte d'entrée potentielle à une endocardite bactérienne. Cette prévention est utile à tout âge et quelle que soit la forme de la CIV, même la plus bénigne. En présence d'une CIV type « maladie de Roger » (c'est-à-dire sans gravité ou retentissement) on a coutume de dire que le patient doit consulter plus souvent son dentiste (tous les 6 mois) que son cardiologue (tous les deux ans ou plus). Avoir une CIV (et plus généralement une malformation cardiaque quelle qu'elle soit) implique d'avoir une dentition parfaite, sans la moindre carie.
En dehors du traitement des infections, la nécessité de recourir à un traitement peut être schématisée comme suit :
- 70 % des CIV ne nécessitent aucun traitement et peuvent mener une vie strictement normale, comportant éventuellement des activités sportives en compétition ;
- 20 % des CIV justifieront d'un traitement médical, habituellement transitoire dans l'attente soit de la fermeture partielle ou complète spontanée de la communication, soit de sa fermeture chirurgicale ;
- 10 % des CIV au plus nécessiteront une intervention de fermeture chirurgicale.
Chez l'adulte, la prise en charge a fait l'objet de la publication de recommandation. Celles, européennes, datent de 2020[10].
Traitement médical
C'est un traitement visant à diminuer les manifestations d'insuffisance cardiaque et d'œdème pulmonaire. Il peut être indiqué chez le nourrisson dans les premiers mois de vie. Il sera éventuellement (mais pas toujours) initié lors d'une courte hospitalisation mais la règle générale est d'éviter le plus possible les hospitalisations (et encore plus les hospitalisations prolongées), et de conduire ce traitement à domicile, avec des médicaments donnés par voie orale, sous couvert d'une surveillance régulière en consultation.
Ce traitement repose sur l'utilisation de la Digoxine (digitalique), d'utilité contestée par certains[Qui ?], de diurétiques (médicaments augmentant la diurèse) et de médicaments vasodilatateurs tels que les inhibiteurs de l'enzyme de conversion de l'angiotensine principalement. On y adjoindra souvent des mesures de puériculture telle que repos et calme, atmosphère tempérée dans la chambre, position surélevée de la tête du berceau, alimentation par un lait enrichi, etc. et patience des parents lors de la prise des biberons qui est d'une lenteur désespérante. On limitera au mieux les contacts avec l'entourage (en particulier les frères ou sœurs scolarisés) pour limiter les risques de contage infectieux. Pour les mêmes raisons, les séjours en crèche sont fortement déconseillés et on ne peut envisager le recours à une gardienne (expérimentée) que si elle ne s'occupe que de votre enfant ou, à la rigueur, d'autres nourrissons du même âge.
Ce n'est que dans les formes graves ou compliquées (par une infection par exemple) que l'on pourra être amené à proposer :
- une alimentation par voie entérale (c'est-à-dire par sonde), éventuellement réalisable à domicile ;
- des mesures transitoires de réanimation telles que assistance respiratoire.
Tout à fait à part se situe le traitement médical proposé aux patients présentant une CIV avec hypertension artérielle pulmonaire fixée. À ce stade, il n'y a habituellement plus d'insuffisance cardiaque et les objectifs du traitement sont de deux ordres :
- diminuer ou freiner l'évolution de la maladie vasculaire pulmonaire à l'aide de traitements vasodilatateurs spécifiques ;
- prévenir ou traiter certaines conséquences de cette maladie: Polyglobulie (excès de globules rouges), thromboses, hypoxie, etc.
Traitement chirurgical
Deux types d'intervention chirurgicales peuvent être envisagées :
- le cerclage (ou banding) de l'artère pulmonaire. Il s'agit d'une intervention « palliative », c'est-à-dire qui vise à limiter de façon transitoire les conséquences de l'anomalie sans corriger celle-ci ;
- la fermeture de la CIV par un patch de péricarde ou de tissu synthétique. C'est l'intervention « curative » et (normalement) définitive.
Leurs places respectives ont fortement évolué au cours du temps et actuellement, dans les pays développés, la plupart des CIV font d'emblée l'objet d'une fermeture par patch, sans mise en place préalable d'un cerclage.
Cerclage de l'artère pulmonaire
Lors de cette intervention, le chirurgien enserre le tronc de l'artère pulmonaire dans un manchon tissulaire afin d'en réduire le diamètre. Ce rétrécissement est à l'origine d'une augmentation de pression dans le ventricule droit et donc d'une diminution du gradient de pression entre les deux ventricules. La quantité de sang passant au travers de la CIV s'en trouve diminuée, ce qui soulage d'une part l'insuffisance ventriculaire gauche, d'autre part l'hypervascularisation pulmonaire.
N.B. : Cette intervention se fait par un abord intercostal entre deux côtes), laissant une cicatrice thoracique gauche, latérale et postérieure, courant entre deux côtes.
- L'intérêt de cette intervention réside dans le fait qu'elle est réalisable à tout âge, qu'elle ne nécessite pas de circulation extra-corporelle ni de mesures de réanimation sophistiquées en per ou post-opératoire.
- Ses inconvénients sont, outre le fait qu'elle ne corrige pas la malformation et qu'elle ne peut donc être que transitoire (sauf cas particulier), la difficulté à « calibrer » correctement le cerclage (ni trop serré ni trop lâche), le risque d'entraver le jeu des valves pulmonaires s'il est placé trop bas, celui d'étrangler une artère pulmonaire s'il est trop haut et enfin le risque de lésion au niveau du tronc pulmonaire lui-même nécessitant une plastie d'élargissement secondairement lors du retrait du cerclage.
Historiquement, cette intervention, faite pour la première fois en 1951 par Muller et Damman[11], était fréquemment pratiquée, faute de mieux, soit parce que les interventions sous CEC n'étaient pas encore envisageable chez l'enfant, soit parce qu'elles nécessitaient qu'il fasse un poids minimum que l'on ne pouvait espérer atteindre à temps. Actuellement, cette intervention reste indiquée dans certaines formes de CIV difficilement accessibles au chirurgien chez le tout-petit et le cerclage reste l'intervention de choix dans les pays en voie de développement qui connaissent les mêmes difficultés d'accès à une circulation extra corporelle.
Fermeture sous CEC
Dans les équipes hautement spécialisées et si nécessaire, cette intervention peut être envisagée sur des nourrissons pesant 4 kg ou moins sans majoration significative des complications per ou post-opératoires[12]. C'est donc l'intervention de choix, faite en première intention dans la grande majorité des CIV.
L'abord se fait par sternotomie médiane (c.a.d ouverture du sternum). L'intervention elle-même se fait sur cœur arrêté, sous circulation extracorporelle (CEC). Le chirurgien doit ouvrir le cœur pour accéder à sa cloison médiane et à la communication. Le mode d'ouverture dépend de la localisation de la ou des CIV. Le plus possible, le chirurgien évite d'ouvrir une paroi ventriculaire (en particulier du ventricule gauche) pour éviter de fragiliser ce muscle toujours en travail. La CIV peut être abordée au travers de la valve tricuspide après ouverture de l'oreillette droite ou au travers des valves pulmonaires après ouverture du tronc pulmonaire. La communication est fermée par une pièce (« patch ») de tissu synthétique fixée par des points de suture sur le versant droit du septum interventriculaire. Le tissu synthétique utilisé est bien toléré et ne présente pas de risque de rejet. Au bout de quelques mois, il sera colonisé par des cellules de l'organisme qui le tapisseront comme elles tapissent le reste du septum (endothélialisation).
Les parents s'interrogent souvent sur « la durée de l'intervention ». En fait, il est plus informatif de savoir que l'enfant « restera au bloc » 2 à 4 heures, même si l'intervention elle-même ne dure qu'une heure environ. Il y a en effet tout un temps de préparation, de mise en route de la CEC avant l'intervention puis tout un temps de surveillance, de « défibrillation du cœur » puis d'arrêt de la CEC après celle-ci. À la sortie du bloc opératoire, l'enfant passera quelques jours en unité de réanimation où seront retirés progressivement tous les « tuyaux » : sonde d'intubation (pour l'assistance respiratoire), drains de redon (pour évacuer les résidus sanguins ou liquidiens dans le thorax), électrodes de stimulation (mises en place préventivement au cas où l'activité électrique du cœur serait perturbée transitoirement en post opératoire).
La cicatrice qui en résulte est impressionnante pour les parents. Outre le confort qu'elle donne au chirurgien pour intervenir sur un organe guère plus gros et aussi délicat qu'un mécanisme d'horlogerie, cette façon d'ouvrir le thorax est la moins douloureuse pour l'enfant en post-opératoire et que c'est celle qui laissera le moins de séquelle (car elle respecte les muscles, les nerfs et les vaisseaux). Habituellement, les nourrissons « cicatrisent bien » et en quelques mois la « balafre » initiale laisse place à une cicatrice discrète, fine ligne légèrement décolorée qui grandit moins vite que le thorax et semble donc diminuer proportionnellement avec le temps. Dans certains cas cependant, il apparaît une déformation sternale (qui le plus souvent s'atténue lors de l'adolescence) et, au lieu de s'estomper, la cicatrice tend à former des excroissances tissulaires (cicatrice chéloïde) en particulier chez les enfants de couleur de peau noire. Une intervention de chirurgie esthétique peut alors être envisagée à l'adolescence.
La durée du séjour hospitalier est habituellement d'une quinzaine de jours ou même moins, à l'issue desquels l'enfant regagne le domicile familial sans traitement ou avec un traitement médical allégé et temporaire.
Cette intervention est unique. Il n'y aura pas besoin de ré-opérer pour changer le patch quand l'enfant grandira. L'enfant pourra mener une vie normale sans traitement, faire du sport y compris en compétition. Les résultats sont, en règle, excellents, avec une mortalité et une morbidité très faible[13].
Traitement par cathétérisme interventionnel
La fermeture par mise en place d'un dispositif obstructif au cours d'un cathétérisme (donc sans chirurgie) est devenue le traitement courant de certaines CIA (communication interauriculaire) ou de la persistance du canal artériel. Cette technique est proposée pour les CIV, en particulier lorsqu'elles siègent dans le septum musculaire, plus difficiles d'accès pour le chirurgien[14]. Sa réalisation est plus délicate pour les CIV périmembraneuses en raison de la proximité des voies de conduction et du risque de survenue d'un bloc atrio-ventriculaire (pouvant nécessiter la mise en place d'un pacemaker)[15].
Devenir des enfants
L'évolution à moyen et long terme est à envisager selon la gravité initiale de la CIV.
CIV de type « maladie de Roger »
La seule précaution à prendre est la prévention de l'endocardite et donc la nécessité de traiter par antibiotiques toute infection bactérienne (les antibiotiques sont inutiles en cas d'infection virale comme une grippe). On a coutume de dire que les personnes présentant une maladie de Roger doivent consulter plus souvent leur dentiste (tous les 6 mois) que leur cardiologue.
Il y a néanmoins lieu de maintenir une surveillance cardiologique régulière, tout particulièrement quand cette CIV siège à proximité des valves aortiques. En effet, dans 5 % des cas environ, une fuite peut apparaître au niveau de ces valves (insuffisance aortique) et l'on envisagera une intervention chirurgicale pour d'une part fermer cette CIV, d'autre part réparer la valves aortique. Cette rare complication est connue sous l'appellation de syndrome de Pezzi-Laubry.
En règle générale, les personnes présentant une maladie de Roger peuvent mener une vie strictement normale et pratiquer les sports de leur choix, y compris en compétition. Entre les deux guerres, un des vainqueurs de la coupe Davis était porteur d'une maladie de Roger.
Les femmes peuvent envisager sans crainte une grossesse, avec un accouchement normal (sans césarienne sauf indication obstétricale) et l'allaitement de leur enfant. Seule précaution, l'accouchement et les suites de couches se feront sous antibiothérapie prophylactique.
Les formes péri-mzembraneuses peuvent se fermer spontanément durant l'enfance, et ce, d'autant que le shunt est petit[16].
CIV ayant fait l'objet d'une intervention de fermeture
En règle générale, cette intervention permet une guérison « complète » et autorise donc une vie tout à fait normale[17].
Quelques limitations, en particulier à l'effort, peuvent subsister lorsque l'intervention ne s'est pas parfaitement déroulée (persistance d'une petite communication résiduelle, trouble de l'activation électrique du cœur...), lorsqu'elle a été un peu tardive (persistance d'un certain degré d'hypertension pulmonaire) ou lorsqu'elle n'a pas pu traiter complètement d'autres anomalies associées.
CIV n'ayant pas pu être opérées (syndrome d'Eisenmenger)
Il s'agit le plus souvent de CIV larges (et qui à ce titre auraient pu justifier une intervention) mais qui s'accompagnent d'une élévation des résistances pulmonaires telle que la fermeture de la CIV ne suffirait pas à les normaliser (hypertension artérielle pulmonaire fixée). On considère que dans ce cas, il vaut mieux ne pas fermer la communication, ce qui risquerait plus d'aggraver l'état du patient que de le soulager.
L'évolution se fait alors vers une diminution puis une inversion du shunt qui devient droit-gauche et s'accompagne de cyanose (Syndrome d'Eisenmenger). Le traitement médical de l'hypertension artérielle pulmonaire passe alors au premier plan. À un stade avancé de l'évolution, une transplantation cœur-poumons peut être parfois envisagée.
Cette forme peut être réellement invalidante. Elle contre-indique la pratique de sports violents et de toute compétition même lorsque le patient apparait asymptomatique dans la vie courante. De même, c'est une des très rares cardiopathies congénitales qui contre-indique formellement la grossesse. Une grossesse précipite l'évolution naturelle de la maladie c'est-à-dire qu'au mieux elle aggraverait l'état de la patiente, au pire elle pourrait être responsable de son décès. Il y aurait également un risque important que la grossesse n'arrive pas à son terme (mort fœtale in utero) ou fasse naitre un enfant très dysmature.
L'évolution naturelle d'une hypertension artérielle pulmonaire fixée secondaire à une CIV, habituellement étalée sur plusieurs décades, autorise par contre le plus souvent une procédure d'adoption.
Prévention de l'endocardite
L'infection de la CIV est une complication classique mais devenue rare.
La prévention par antibiothérapie systématique avant certains soins (en particulier dentaire) n'est plus en vigueur pour la majorité des cas aux États-Unis. Elle reste toutefois prescrite dans les premiers mois après une intervention de fermeture (qu'elle soit par chirurgie ou par voie endovasculaire)[18] et nécessaire pour certaines interventions chez les personnes à risque modéré ou fort d'endocardite[19].
Une bonne hygiène buccodentaire reste nécessaire dans tous les cas.
Conseil génétique
La plupart des CIV peuvent être considérées comme un accident sans lendemain (« un défaut de finition ») dont le risque de récidive est proche de celui observé dans la population générale. À ce titre, elles ne justifient pas un conseil génétique.
Un conseil génétique, non lié à la présence de la CIV, mais de l'anomalie chromosomique qui l'accompagne, se justifiera dans les situations suivantes :
- CIV postérieure liée à l'existence d'une trisomie 21 ;
- CIV sous-aortique survenant dans le cadre d'une microdélétion 22q11 ;
- CIV observée au sein d'un syndrome polymalformatif révélateur d'une trisomie 13, d'une trisomie 18 ou d'une autre anomalie génétique révélée par le caryotype fœtal ou néonatal.
Articles connexes
- Communication interauriculaire
- Cardiopathie congénitale
- Syndrome de Bland-White-Garland
- Insuffisance cardiaque
- Malformation congénitale
- Sténose de l'artère pulmonaire
Notes et références
Notes
Références
- « Les shunts », sur precisdanesthesiecardiaque.ch
- (en) Penny DJ, Vick JW, « Ventricular septal defect » Lancet, 2011;377:1103-1112
- (en) Roguin N, Du ZD, Barak M, Nasser N, Hershkowitz S, Milgram E, « High prevalence of muscular ventricular septal defect in neonates » J Am Coll Cardiol. 1995;26:1545-1548
- (en) Jenkins KJ, Correa A, Feinstein JA et al. « Noninherited risk factors and congenital cardiovascular defects: current knowledge: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics » Circulation 2007;115:2995-3014
- (en) Axt-Fliedner R, Schwarze A, Smrcek J, Germer U, Krapp M, Gembruch U, « Isolated ventricular septal defects detected by color Doppler imaging: evolution during fetal and first year of postnatal life » Ultrasound Obstet Gynecol. 2006;27:266-273
- (en) Kilner PJ, Geva T, Maemmerer H, Trindade PT, Schwitter J, Webb GD, « Recommendations for cardiovascular magnetic resonance in adults with congenital heart disease from the respective working groups of the European Society of Cardiology » Eur Heart J. 2010;31:794-805
- Nadas AS Pediatric cardiology 2e édition 1964 - W.B Saunders Company
- Nouaille J, Gautier M, Lucet P, Mercier JN, Kachaner J Les communications interventriculaires du nourrisson et de l'enfant. À propos de 500 observations. I. Étude clinique et problèmes thérapeutiques. Arch Mal Cœur 1967;60: 1138
- Lopez L, Houyel L, Colan SD et al. Classification of ventricular septal defects for the Eleventh Iteration of the International Classification of Diseases-striving for consensus: a report from the International Society for Nomenclature of Paediatric and Congenital Heart Disease, Ann Thorac Surg, 2018;106:1578–1589
- Baumgartner H, De Backer J, Babu-Narayan SV et al. 2020 ESC Guidelines for the management of adult congenital heart disease: The Task Force for the management of adult congenital heart disease of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Adult Congenital Heart Disease (ISACHD), European Heart Journal, 2021;42:563–645
- (en) Muller WH, Dammann JF, « Treatment of certain congenital malformations of the heart by the creation of pulmonic stenosis to reduce pulmonary hypertension and excessive pulmonary blood flow: A preliminary report » Surgery Gynecol Obstet. 1952;95:213
- (en) Kogon B, Butler H, Kirshbom P, Kanter K, McConnell M, « Closure of symptomatic ventricular septal defects: how early is too early? » Pediatr Cardiol. 2008;29:36-9
- (en) Scully BB, Morales DLS, Zafar F, McKenzie ED, Fraser CD, Heinle JS, « Current expectations for surgical repair of isolated ventricular septal defects » Ann Thorac Surg. 2010;89:544-549
- (en) Lim DS, Forbes TJ, Rothman A, Lock JE, Landzberg MJ, « Transcatheter closure of high-risk muscular ventricular septal defects with the CardioSEAL occluder: initial report from the CardioSEAL VSD registry » Catheter Cardiovasc Interv. 2007;70:740-744
- (en) Carminati M, Butera G, Chessa M et al. Investigators of the European VSD Registry. « Transcatheter closure of congenital ventricular septal defects: results of the European Registry » Eur Heart J. 2007:28):2361-8
- Miyake T, Shinohara T, Fukuda T, Ikeoka M, Takemura T, Spontaneous closure of perimembranous ventricular septal defect after school age, Pediatr Int, 2008;50:632–635
- Meijboom F, Szatmari A, Utens E, Deckers JW, Roelandt JR, Bos E, Hess J, Long-term follow-up after surgical closure of ventricular septal defect in infancy and childhood, J Am Coll Cardiol, 1994;24:1358–1364
- (en) Wilson W, Taubert KA, Gewitz M et al. « Prevention of infective endocarditis: guidelines from the American Heart Association: a guideline from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group » Circulation 2007;116:1736-1754
- « Prévention de l'Endocardite », Esculape.com, mise à jour en 2010. Sources : « Cinquième Conférence De Consensus - 27 mars 1992 - Paris » et « AHA Guidelines - 2007 »
 Portail du handicap
Portail du handicap  Portail de la médecine
Portail de la médecine