Citrate synthase
La citrate synthase (CS) est une acyltransférase qui catalyse la réaction :
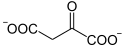
- acétyl-CoA + H2O + oxaloacétate → citrate + CoA.
| Citrate synthase | ||
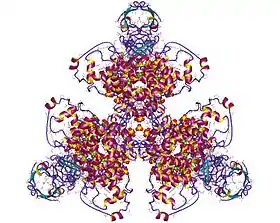
 Structure d'une citrate synthase d’Acetobacter complexée avec l'oxaloacétate (en magenta) et de la carboxyméthyl-déthia-coenzyme A (en jaune, inhibiteur) résistante à l'acidité (PDB 1CSI[1]) | ||
| Caractéristiques générales | ||
|---|---|---|
| Symbole | CS | |
| N° EC | 2.3.3.1 | |
| Homo sapiens | ||
| Locus | 12q13.3 | |
| Masse moléculaire | 51 712 Da[2] | |
| Nombre de résidus | 466 acides aminés[2] | |
| Entrez | 1431 | |
| HUGO | 2422 | |
| OMIM | 118950 | |
| UniProt | O75390 | |
| RefSeq (ARNm) | NM_004077.2 | |
| RefSeq (protéine) | NP_004068.2 | |
| Ensembl | ENSG00000062485 | |
| Liens accessibles depuis GeneCards et HUGO. | ||
Cette enzyme intervient à la 1re étape du cycle de Krebs, où elle catalyse une réaction irréversible qui engage cette voie métabolique[3] : la condensation du résidu acétyle de l'acétyl-CoA sur l'oxaloacétate pour former du citrate en libérant la coenzyme A ; l'oxaloacétate est régénéré après un tour complet du cycle de Krebs. Elle est présente chez presque tous les êtres vivants connus. Chez les eucaryotes, elle est active dans la matrice mitochondriale, où se déroule le cycle de Krebs, mais est codée dans le génome nucléaire, traduite par des ribosomes cytosoliques, et enfin transportée à l'intérieur des mitochondries à travers la membrane mitochondriale interne. Elle est couramment utilisée en laboratoire comme marqueur enzymatique pour mesurer la quantité de mitochondries intactes.
Il existe deux isoenzymes catalysant cette réaction avec une stéréospécificité opposée :
- la citrate (Si)-synthase (EC ) est l'enzyme la plus répandue, avec une stéréospécificité Si[4] ;
- la citrate (Re)-synthase (EC ) est présente chez certains organismes anaérobies ; il s'agit d'une isoforme inactivée par l'oxygène ayant une stéréospécificité Re.
| N° EC | EC |
|---|---|
| N° CAS |
| IUBMB | Entrée IUBMB |
|---|---|
| IntEnz | Vue IntEnz |
| BRENDA | Entrée BRENDA |
| KEGG | Entrée KEGG |
| MetaCyc | Voie métabolique |
| PRIAM | Profil |
| PDB | RCSB PDB PDBe PDBj PDBsum |
| N° EC | EC |
|---|---|
| N° CAS |
| IUBMB | Entrée IUBMB |
|---|---|
| IntEnz | Vue IntEnz |
| BRENDA | Entrée BRENDA |
| KEGG | Entrée KEGG |
| MetaCyc | Voie métabolique |
| PRIAM | Profil |
| PDB | RCSB PDB PDBe PDBj PDBsum |
Structure et mécanisme
L'oxaloacétate est le premier substrat à se lier à l'enzyme, ce qui induit un changement conformationnel créant un site de liaison pour l'acétyl-CoA. Ces deux molécules se condensent alors pour former de la citryl-CoA, ce qui induit un second changement conformationnel au cours duquel la liaison thioester est hydrolysée, en libérant la coenzyme A[5].
+ acétyl-CoA + H2O → CoA + 
Oxaloacétate Citrate
La structure de la citrate synthase d’Acetobacter a été étudiée par cristallographie aux rayons X[1]. Cette enzyme est formée de 437 résidus d'acides aminés répartis sur deux sous-unités comprenant chacune 20 hélices α. Ces hélices α constituent environ 75 % de la structure tertiaire de l'enzyme, le reste étant des extensions irrégulières de cette structure hormis un petit feuillet β de 13 résidus. Le site actif est situé dans une fente entre ces deux sous-unités. Il contient deux sites de liaison : l'un est réservé au citrate ou à l'oxaloacétate et l'autre à la coenzyme A.
Le site actif contient trois résidus clés — l'His274, l'His320 et l'Asp375 — qui interagissent avec les substrats de manière très spécifique :
 Site actif de la citrate synthase de coq bankiva, montrant les résidus impliqués dans le mécanisme réactionnel de l'enzyme (PDB 1CSI[1]).
Site actif de la citrate synthase de coq bankiva, montrant les résidus impliqués dans le mécanisme réactionnel de l'enzyme (PDB 1CSI[1]).
Ces trois résidus forment une triade catalytique qui assure la crotonisation de l'oxaloacétate et de l'acétyl-CoA. La réaction commence avec la déprotonation de l'atome de carbone α de l'acétyl-CoA sous l'effet de la charge électrique négative de la chaîne latérale du résidu Asp375. Cette déprotonation conduit à la formation d'un anion énolate, qui est à son tour protoné par la chaîne latérale du résidu His274 pour donner un énol. Le doublet non liant de l'atome d'azote ε de ce résidu His274 agit en déprotonant l'hydroxyle de l'énol formé à l'étape précédente, ce qui redonne un anion énolate. Ce dernier amorce une attaque nucléophile sur l'atome de carbone du carbonyle de l'oxaloacétate qui déprotone l'atome d'azote ε du résidu His320. Il en résulte l'addition nucléophile des deux substrats, conduisant à la citryl-CoA. Le résidu His320 déprotoné agit en déprotonant une molécule d'eau, ce qui déclenche l'hydrolyse de la liaison thioester de la citryl-CoA. L'un des doublets non liants de l'oxygène agit dans une attaque nucléophile sur l'atome de carbone du carbonyle de la citryl-CoA, ce qui forme un intermédiaire tétraédrique qui conduit à expulser la coenzyme A lorsque le carbonyle se reconstitue[6].
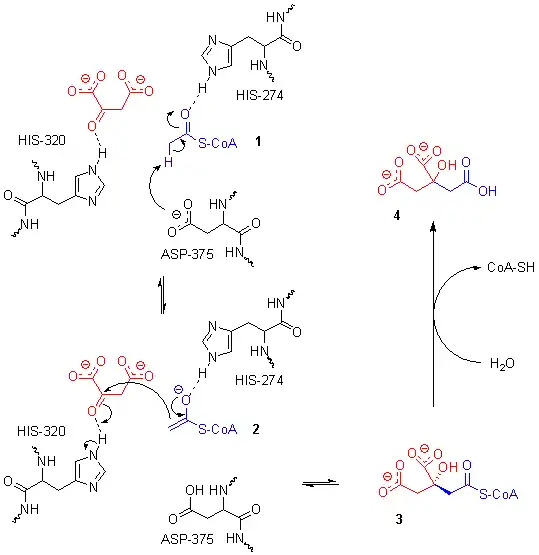
Mécanisme réactionnel de la citrate synthase.
Inhibition
La citrate synthase est inhibée par une valeur élevée des ratios de concentration [ATP] / [ADP], [acétyl-CoA] / [CoA] et [NADH] / [NAD+], qui sont des indicateurs de charge énergétique élevée dans la cellule. Elle est également inhibée par la succinyl-CoA, dont la molécule ressemble à celle de l'acétyl-CoA et agit comme inhibiteur compétitif. Le citrate inhibe également la réaction, illustrant un mécanisme de rétro-inhibition par le produit de réaction.
L'inhibition de la citrate synthase par des analogues de l'acétyl-CoA est également abondamment documentée et a été utilisée pour démontrer l'existence d'un site actif unique. Ces expériences ont montré que ce site actif oscille entre deux formes, qui contribuent pour l'une à l'activité ligase et pour l'autre à l'activité hydrolase de cette enzyme[5].
La citrate synthase serait régie par un mode de régulation allostérique de type morphéine[7].
Notes et références
- (en) Ken C. Usher, S. James Remington, David P. Martin et Dale G. Drueckhammer, « A Very Short Hydrogen Bond Provides Only Moderate Stabilization of an Enzyme-Inhibitor Complex of Citrate Synthase », Biochemistry, vol. 33, no 25, , p. 7753-7759 (PMID 8011640, DOI 10.1021/bi00191a002, lire en ligne)
- Les valeurs de la masse et du nombre de résidus indiquées ici sont celles du précurseur protéique issu de la traduction du gène, avant modifications post-traductionnelles, et peuvent différer significativement des valeurs correspondantes pour la protéine fonctionnelle.
- (en) G. Wiegand et S. J. Remington, « Citrate Synthase: Structure, Control, and Mechanism », Annual Review of Biophysics and Biophysical Chemistry, vol. 15, , p. 97-117 (PMID 3013232, DOI 10.1146/annurev.bb.15.060186.000525, lire en ligne)
- (en) « Re, Si », Compendium of Chemical Terminology [« Gold Book »], IUPAC, 1997, version corrigée en ligne : (2006-), 2e éd.
- (en) Ernst Bayer, Barbara Bauer et Hermann Eggerer, « Evidence from Inhibitor Studies for Conformational Changes of Citrate Synthase », The FEBS Journal, vol. 120, no 1, , p. 155-160 (PMID 7308213, DOI 10.1111/j.1432-1033.1981.tb05683.x, lire en ligne)
- (en) David L. Nelson et Michael M. Cox, Lehninger Principles of Biochemistry, W. H. Freeman, , 4e éd., 1100 p. (ISBN 978-0-7167-4339-2)
- (en) Trevor Selwood et Eileen K. Jaffe, « Dynamic dissociating homo-oligomers and the control of protein function », Archives of Biochemistry and Biophysics, vol. 519, no 2, , p. 131-143 (PMID 22182754, PMCID 3298769, DOI 10.1016/j.abb.2011.11.020, lire en ligne)
 Portail de la biochimie
Portail de la biochimie  Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire