Coarctation de l'aorte
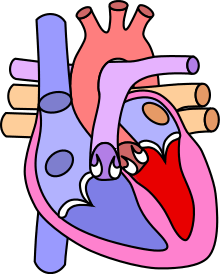
La coarctation de l'aorte est un rétrécissement congénital de l'aorte, situé juste en dessous de l'émergence de l'artère subclavière gauche, marquant le début de l'aorte thoracique descendante.
Historique
L'anomalie a été décrite dès 1760 par l'anatomiste prussien Johann Friedrich Meckel[1]. Les premières interventions chirurgicales datent des années 1940, les premières angioplasties des années 1980
Anatomie et physio-pathologie
Elle n'est pas, stricto sensu, une cardiopathie congénitale, ne concernant pas le cœur, bien que très souvent traitée dans ce type de pathologie.
Le canal artériel (Ductus Arterios Botalli) est un vaisseau permettant le passage du sang de l'artère pulmonaire vers l'aorte (distalement de l'artère sous-clavière gauche) durant la vie fœtale. À la naissance, il involue et se transforme en ligament artériel. La coarctation serait secondaire à la persistance de tissu de ce canal artériel (tissu musculaire et élastique) dans la paroi de l'aorte[2].
La zone entre l'émergence de l'artère sous-clavière gauche et la première paire d'artères intercostales est nommée l'isthme aortique. La coarctation peut se situer avant ou après le ligament artériel, ainsi on distingue la coarctation pré-ductale ou post-ductale.
Il existe une augmentation de la pression en amont du rétrécissement et une diminution en aval. Une collatéralité va se former, permettant de faciliter le passage du sang vers le bas. Cette collatéralité se développe, en particulier, au niveau des artères intercostales qui augmentent de volume, devenant tortueuse et pouvant imprimer leur forme sur leur côte respective[2].
L'aorte, en amont du rétrécissement, peut également se dilater jusqu'à former un anévrisme qui peut se compliquer d'une dissection aortique (fissuration de la paroi aortique de manière longitudinale) gravissime[3].
Cette anomalie s'accompagne dans 90 % des cas de cardiopathie congénitale. Une malformation extra-cardiaque est retrouvée dans 13 % des cas. Cette anomalie est retrouvée entre 10 % et 35 % des syndromes de Turner. La bicuspidie valvulaire aortique est fréquente[2] : anomalie de la valve aortique constitués de deux feuillets au lieu de trois.
Diagnostic
Anténatal

Le diagnostic anténatal est possible mais se heurte à une difficulté majeure: en raison de la participation du tissu de canal artériel à sa formation, l'étude la plus stricte et dans des conditions optimum de la crosse de l'aorte peut être strictement normale et pourtant quelques jours après la coarctation apparaît; mais il existe parfois des signes.
Aspects échographiques avant la naissance
Le signe direct est la mise en évidence du rétrécissement mais est en pratique assez difficile à détecter.
Les signes indirects sont une augmentation du ventricule droit et de l'artère pulmonaire sont des signes plus faciles à mettre en évidence. Près de deux tiers des coarctations de l'aorte se manifesteraient in utero par ce signe; mais il existe une augmentation physiologique de la taille du ventricule droit en fin de grossesse et la présence d'une communication inter-ventriculaire peut masquer ce signe.
D'après Allan[4], les signes suivants permettraient de suspecter une coarctation de l'aorte:
- En cas communication inter-ventriculaire, la présence d'un shunt gauche-droit prédominant au doppler couleur.
- Le diamètre moyen de l'aorte est plus petit alors que le diamètre moyen de l'artère pulmonaire est plus grand entraînant un rapport Aorte/Artère pulmonaire bien en dessous des normes est un signe fiable.
- Le signe le plus fiable serait l'hypoplasie de l'arc aortique avec un flux sanguin inférieur au cinquième percentile au niveau de l'isthme aortique.
Le principal diagnostic différentiel à éliminer est la sténose pulmonaire, mais dans ce cas l'artère pulmonaire a un diamètre réduit alors que dans la coarctation son diamètre augmente.
Gestion de la grossesse
En raison de la forte association avec un syndrome de Turner, un caryotype pourra être proposé aux parents. Un suivi régulier de l'évolution de la coarctation au cours de la grossesse est indispensable en raison de l'évolution possible de cette pathologie in utero, et incite à la recherche de signes d'insuffisance cardiaque. Cette pathologie nécessite rarement un accouchement prématuré. La naissance devra se faire dans une maternité disposant d'un service de cardiologie pédiatrique.
Prise en charge du nouveau-né
Postnatal
En cas d'hypertension chez un individu jeune (<30-40 ans), il faudra toujours rechercher le pouls fémoral. En effet, en cas d'absence de celui-ci, il faudra penser à une coarctation de l'aorte. Il faut savoir que la plupart des coarctations aortique ne sont pas diagnostiquées pendant la grossesse ; vu les difficultés citées plus haut. Or à la naissance l'enfant peut paraitre en parfaite santé et même rentrer chez lui. La décompensation n'arrivera donc pas à la naissance mais quelques jours après, lorsque les résistances pulmonaires sont au plus bas.
Traitement
Dans une coarctation simple, l'âge idéal de l'intervention est aux environs de 1 an pour pratiquer l'intervention de Crafoord. Pratiquée trop tôt, le risque de coarctation augmente, pratiquée plus tard, le risque de voir l'hypertension artérielle persister est important.
En fait, la date de l'intervention dépend de la possibilité du ventricule gauche d'assurer la circulation systémique.
L'intervention préférentielle est l'intervention de Crafoord consistant en la résection chirurgicale de la partie de l'aorte ayant la coarctation avec anastomose termino-terminale (suture bout-à bout). Si la coarctation est étendue, un patch d'agrandissement peut s'avérer nécessaire[5].
Un traitement alternatif est l'angioplastie : un ballon est introduit par l'artère fémorale et positionné, sous contrôle radiologique, au niveau de la coarctation. Il est alors gonflé, entraînant la dilatation de cette dernière. Elle a pour avantage de ne pas nécessiter d'intervention chirurgicale et a de bons résultats initiaux[6]. Elle comporte néanmoins un risque de formation d'un anévrisme à long terme[7]. Le geste peut être complété par la mise en place d'un stent[8].
Devenir
La récidive de la coarctation après traitement est possible et est d'autant plus fréquente que la prise en charge a été précoce (chez le nouveau-né)[9]. Elle peut être due à une réparation incomplète (ablation imparfaite de la coarctation) ou à une cicatrice sténosante sur l'aorte et peut nécessiter parfois une réintervention.
Le risque d'apparition d'une hypertension artérielle est majorée à l'âge adulte[10] sans que cela ait un rapport avec une récidive surtout si le traitement est tardif (chez l'adulte)[11]. Les raisons n'en sont pas claires : il semble exister une dysfonction endothéliale[12] ainsi qu'une modification de la réactivité artérielle[13] après réparation de la coarctation. Cela impose donc une surveillance régulière et la mise en route, si besoin, d'un traitement antihypertenseur. Le plus grand risque de survenue de maladies cardio-vasculaires semble plus être en rapport avec les facteurs de risque (dont l'hypertension artérielle) que du fait de la coarctation elle-même[14].
Conseil génétique
En l'absence de maladie à transmission héréditaire, le risque d'avoir un autre enfant atteint de cette pathologie est de 2 % mais passe à 6 % si deux enfants sont atteints. En cas d'un parent porteur de cette pathologie, le risque est de entre 2,7 % à 4 %[15].
Références
- Jarcho S, Coarctation of the aorta (Meckel 1750, Paris 1791), Am J Cardiol, 1961;7:844–852
- Perloff JK, The variant associations of aortic isthmic coarctation, Am J Cardio, 2010;106:1038-1041
- Doyle L, Coarctation of the aorta with hemopericardium, 1991:46;268–269
- Coarctation of the aorta, L. D. Allan, King's College Hospital, UK Conference: 13th World Congress on Ultrasound in Obstetrics and Gynecology, Paris, France, 31 August 2003 to 4 September 2003.
- Gaca AM, Jaggers JJ, Dudley LD, Bisset III GS, Repair of congenital heart disease: a primer—part 2, Radiology, 2008;248:44–60
- Rao PS, Galal O, Smith PA et Als. Five- to nine-year follow-up results of balloon angioplasty of native aortic coarctation in infants and children, J Am Coll Cardiol, 1996;27:462–70
- Cowley CG, Orsmond GS, Feola P et Als. Long-term, randomized comparison of balloon angioplasty and surgery for native coarctation of the aorta in childhood, Circulation 2005;111:3453–6
- Forbes J, Garekar S, Amin Z et al. Procedural results and acute complications in stenting native and recurrent coarctation of the aorta in patients over 4 years of age: a multi-institutional study, Catheter Cardiovasc Interv, 2007;70:276–285
- Younoszai AK, Reddy VM, Hanley FL et Als. Intermediate term follow-up of the end-to-side aortic anastomosis for coarctation of the aorta, Ann Thorac Surg, 2002;74:1631–4.
- Hager A, Kanz S, Kaemmerer H, Schreiber C, Hess J, Coarctation long-term assessment (CoALA): significance of arterial hypertension in a cohort of 404 patients up to 27 years after surgical repair of isolated coarctation of the aorta, even in the absence of restenosis and prosthetic material, J Thorac Cardiovasc Surg, 2007;134:738–745.e732
- Seirafi PA, Warner KG, Geggel RL et Als. Repair of coarctation of the aorta during infancy minimizes the risk of late hypertension, Ann Thorac Surg, 1998;66:1378–82
- Brili S, Tousoulis D, Antoniades C, Aggeli C, Roubelakis A, Papathanasiu S, Stefanadis C, Evidence of vascular dysfunction in young patients with successfully repaired coarctation of aorta, Atherosclerosis, 2005;182:97–103
- Gardiner HM, Celermajer DS, Sorensen KE, Georgakopoulos D, Robinson J, Thomas O, Deanfield JE, Arterial reactivity is significantly impaired in normotensive young adults after successful repair of aortic coarctation in childhood, Circulation, 1994;89:1745–1750
- Roifman I, Therrien J, Ionescu-Ittu R, Pilote L, Guo l, Kotowycz MA, Martucci G, Marelli AJ, Coarctation of the aorta and corony artery disease: fact or fiction?, Circulation, 2012;126:16–21
- Nora JJ, Nora AH. Update on counselling the familly with a first-degree relative with a congenital heart defect Am J Med Genet 1988;29;137-142

