Syndrome de Pfeiffer
Le syndrome de Pfeiffer est une craniosynostose en rapport avec une mutation du gène FGPR. Cette mutation du gène FGPR est responsable d'autres craniosynostose regroupées sous le nom de craniosynostose FGPR dépendante. Les sutures du crâne qui fusionnent dans cette maladie sont les sutures coronales avec parfois sagittales. Le syndrome de Pfeiffer comprend différents types en fonction du type du gène FGPR.
| Spécialité | Rhumatologie |
|---|
| CISP-2 | A90 |
|---|---|
| CIM-10 | Q87.0 |
| CIM-9 | 755.55 |
| OMIM | 101600 |
| DiseasesDB | 32145 |
| MeSH | D000168 |
![]() Mise en garde médicale
Mise en garde médicale
Autres noms de la maladie
- Acrocéphalosyndactylie type V
- Syndrome de Noack
- Dysostose craniofaciale et dermatologique
Étiologie
Certaines études suggèrent que l'âge paternel augmenterait le risque de cette pathologie[1],[2].
Le syndrome de Pfeiffer (nl) résulte de deux mutations possibles :
- mutation du gène FGFR1 ou fibroblast growth factor receptor-1 localisé sur le locus 8p11.2-p11.1 du chromosome 8 ;
- mutation du gène FGFR2 ou fibroblast growth factor receptor-2 localisé sur le locus q26 du chromosome 10.
Il n'existe pas d'équivalence entre le type de mutation du gène (FGFR1 ou FGFR2) et la classification en types I, II ou III, qui sont cliniques, c'est-à-dire établis par classification des symptômes :
- le type I peut être causé par des mutations soit dans le gène FGFR1, soit dans le gène FGFR2,
- les types II et III sont causés par des mutations dans le gène FGFR2, mais ne sont pas causés par des mutations dans le gène FGFR1.
Incidence
1 sur 100 000 naissances.
Description
Le médecin allemand Rudolf Pfeiffer (nl) reprend et publie en 1964 la description clinique d'une famille, où le syndrome d'Apert avait été diagnostiqué de façon erronée : il est l'éponyme du nouveau syndrome qu'il décrit[3].
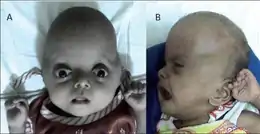
Syndrome de Pfeiffer type I
Le plus courant. Les enfants conservent une intelligence normale, la face est assez caractéristique. Le pouce et le gros orteil sont larges et déviés. Les doigts sont plus ou moins courts. Parfois il existe une perte d’audition.
Syndrome de Pfeiffer type II et type III
Retard mental fréquent. Déformation très importante du crâne avec protrusion des yeux importante pouvant empêcher la fermeture des paupières. Les atteintes des mains et des pieds sont pareilles au type I.
Diagnostic
Le diagnostic est essentiellement clinique :
Anténatal
Dans les familles à risque, une recherche de la mutation est possible par prélèvement de trophoblaste ou par amniocentèse. La découverte fortuite (en cas de mutation de novo) par échographie est possible.
Différentiel
Le diagnostic différentiel est celui d’une craniosynostose.
Mode de transmission
Conseil génétique
Une enquête familiale est indispensable.
Notes et références
- (en) Glaser RL, Jiang W, Boyadjiev SA, Tran AK, Zachary AA, Van Maldergem L, Johnson D, Walsh S, Oldridge M, Wall SA, Wilkie AO, Jabs EW., « Paternal origin of FGFR2 mutations in sporadic cases of Crouzon syndrome and Pfeiffer syndrome », Am J Hum Genet, vol. 66, no 3, , p. 768-777 (PMID 10712195, lire en ligne)
- (en) Goriely A, Wilkie AO., « Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease », Am J Hum Genet, vol. 90, no 2, , p. 175-200. (PMID 22325359, DOI 10.1016/j.ajhg.2011.12.017, lire en ligne)
- (en) « Article « Syndrome de Pfeiffer » », sur Who Named It?
Liens externes
- Fiche sur Orphanet, site en français de renseignement sur les maladies rares, les services de référence, les associations, et les médicaments orphelins.
- (en)Fiche OMIM, site en anglais incontournable pour les maladies génétiques.
- (en) ghr.nlm.nih.gov, site en anglais de vulgarisation sur la génétique et les effets des variations génétiques sur la santé humaine
 Portail de la médecine
Portail de la médecine  Portail du handicap
Portail du handicap