Microscopie STED
La microscopie STED (stimulated-emission-depletion ou déplétion par émission stimulée)[1] est une microscopie de fluorescence à balayage dont l'illumination est mise en forme pour dépasser la limite de résolution imposée par la diffraction. Cette limite est dépassée en utilisant l'émission stimulée pour éliminer la fluorescence dans les régions extérieures de la réponse impulsionnelle optique d'excitation. La largeur de la zone centrale pouvant encore émettre de la lumière est ajustée grâce à la saturation de la fluorescence.
Cette technique permet d'atteindre une résolution de 2,4 nm en champ lointain[2].
Le , l'Allemand Stefan W. Hell a été récompensé par le prix Nobel de chimie, en compagnie de Eric Betzig et William Moerner (États-Unis), pour la mise au point de la microscopie STED.
Principe : étouffer l'excitation des molécules périphériques
Les bases
Le microscope STED est basé sur un microscope à fluorescence confocal à balayage. Pour mieux comprendre le principe du STED, il est utile de réviser les principes de fluorescence, émission stimulée et microscopie confocale.
Fluorescence et émission stimulée
Pour marquer les structures d'intérêt dans les échantillons qu'on veut observer avec la microscopie STED on utilise des colorants fluorescents ou fluorophores, « excités » à une certaine longueur d'onde: par absorption d'un photon ils sont transférés dans un état plus énergétique. Ils retournent à l’état fondamental par émission spontanée d'un autre photon de longueur d'onde plus large (lumière d'une autre couleur) : c'est la lumière de fluorescence qui rend les structures d'intérêt visibles.
Un fluorophore excité peut également retourner par un autre procédé : l'émission stimulée. Un photon illuminant un fluorophore excité provoque le retour de ce fluorophore à l'état fondamental et l'émission d'un photon, identique au photon d'illumination. L'émission spontanée ne peut donc plus avoir lieu, car le fluorophore n'est plus dans l'état excité. La lumière de fluorescence n'est alors pas observée : ce procédé permet donc d'« éteindre » la fluorescence par émission stimulée. La lumière de fluorescence spontanée et la lumière émise par émission stimulée peuvent être séparées, par exemple par des filtres colorés.
La microscopie fluorescente à balayage laser
Dans un microscope à balayage laser, l'illumination n'est pas plein champ : l’échantillon est balayé par un faisceau laser focalisé en un petit point dans l’échantillon. La fluorescence de ce point est détectée. Par balayage du point focal, une image est construite point par point. La taille du point focal détermine la finesse des détails qui peuvent être résolus. À cause de la diffraction, la taille du point focal est limitée à un rayon minimum. Il est impossible de focaliser un laser sur un point plus petit qu’à peu près la moitié de la longueur d’onde.
Principe du microscope STED
Le moyen sans doute le plus direct pour affiner le point focal de fluorescence est d'inhiber sélectivement la fluorescence dans ses parties extérieures. Si ceci est appliqué à un faisceau limité par la diffraction, on peut s'attendre à ce que la barrière de diffraction soit dépassée puisqu'un point focal plus petit signifie résolution spatiale augmentée.

Dans ce but l’échantillon n’est pas seulement illuminé avec le faisceau d’excitation focalisé (figure de gauche) mais aussi avec un faisceau STED" (impulsion de déplétion) auquel on donne le profil d’un anneau dans l’échantillon (figure du milieu). La longueur d’onde de ce faisceau est choisie de façon qu’il induise l’émission stimulée. Au centre, là où le faisceau d’excitation a son intensité maximale, l'intensité du faisceau STED est minimum et n’y influence pas les fluorophores qui restent lumineux (figure de droite). Par contre, il éteint les fluorophores périphériques par émission stimulée.
L'émission stimulée est donc l'une des clés du STED. Cependant, le STED par lui-même ne pourrait vraiment dépasser la limite de diffraction puisque les faisceaux avec lesquels travaille le STED sont eux-mêmes limités par la diffraction. Le point crucial pour dépasser cette limite de diffraction réside dans le lien non linéaire entre la fluorescence et l'excitation (saturation de l'inhibition de fluorescence). La taille de la tache lumineuse résultante diminue avec l’augmentation de l’intensité du faisceau STED, ce qui améliore la résolution; la résolution atteignable est théoriquement illimitée[3].
Applications
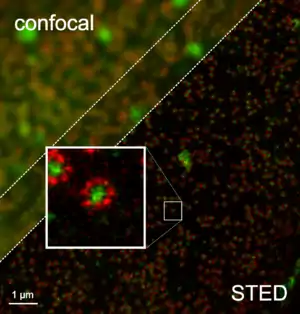
.jpg.webp)
En tant que technique basée sur l'imagerie de fluorescence, la microscopie STED résout le problème de manque de contraste rencontré dans les échantillons biologiques comme les cellules. Les molécules fluorescentes sont liées aux zones d'intérêt via des anticorps ou encore codées génétiquement. Par exemple, on peut marquer spécifiquement les mitochondries. L'observation de fluorescence indique alors la présence de mitochondries (imagerie fonctionnelle). À l'instar du microscope confocal, le microscope STED nécessite donc l'utilisation de marqueurs.
Contrairement à la microscopie électronique il n’est pas nécessaire d'utiliser une enceinte à vide, ni de réaliser des coupes fines. On peut ainsi observer des cellules vivantes[4] et des observations in vivo sont aussi possibles[5].
Contrairement à la microscopie à sonde locale, la microscopie STED est une technique à champ lointain. Elle n’est donc pas limitée à l’observation des surfaces. On peut par exemple voir l’intérieur des cellules et des tissus. L’observation de processus dynamiques est aussi possible[6],[7].
Des microscopes STED sont vendus depuis 2007 par Leica Microsystems.
Références
- Hell, S.W., et al. (1994). Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 19, 780-782
- Wildanger, D., B. R. Patton, H. Schill, L. Marseglia, J. P. Hadden, S. Knauer, A. Schönle, J. G. Rarity, J. L. O’Brien, S. W. Hell, J. M. Smith, « Solid Immersion Facilitates Fluorescence Microscopy with Nanometer Resolution and Sub-Ångström Emitter Localization », Advanced Materials, vol. 24, , p. 309-313 (DOI 10.1002/adma.201203033)
- Stefan W. Hell, « Far-Field Optical Nanoscopy », Science, vol. 316, , p. 1153-1158 (DOI 10.1126/science.1137395)
- Volker Westphal, Silvio O. Rizzoli, Marcel A. Lauterbach, Dirk Kamin, Reinhard Jahn, Stefan W. Hell, « Video-Rate Far-FieldOptical Nanoscopy Dissects Synaptic Vesicle Movement », Science, vol. 320, , p. 246-249 (ISSN 0036-8075, DOI 10.1126/science.1154228)
- Sebastian Berning, Katrin I. Willig, Heinz Steffens, P. Dibaj, Stefan W. Hell, « Nanoscopy in a Living Mouse Brain », Science, vol. 335, , p. 551 (ISSN 0036-8075, DOI 10.1126/science.1215369)
- Volker Westphal, Marcel A. Lauterbach, Angelo Di Nicola, Stefan W. Hell, « Dynamic far-field fluorescence nanoscopy », New Journal of Physics, vol. 9, , p. 435. (DOI 10.1088/1367-2630/9/12/435)
- (en) Marcel A. Lauterbach, Chaitanya K. Ullal, Volker Westphal et Stefan W. Hell, « Dynamic Imaging of Colloidal-Crystal Nanostructures at 200 Frames per Second », Langmuir, vol. 26, , p. 14400-14404 (ISSN 0743-7463, DOI 10.1021/la102474p, lire en ligne)
 Portail de la physique
Portail de la physique  Portail de l’optique
Portail de l’optique  Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire