Loi de Kasha
Appelée ainsi d'après le spectroscopiste américain Michael Kasha, la loi de Kasha est un principe photochimique stipulant qu'après l'absorption d'un photon par une molécule dans l'état fondamental et le peuplement résultant des états excités, l'émission radiative qui s'ensuit (fluorescence ou phosphorescence) se fait depuis l'état excité de plus basse énergie. Il s'agit d'une règle empirique basée sur le fait que les processus de relaxation de l'énergie absorbée depuis les états excités, processus correspondant pour l'essentiel à une relaxation vibrationnelle intramoléculaire (RVI), sont bien plus rapides (de l'ordre de grandeur de ceux d'absorption radiative soit entre 10-15 et 10-12 s) que ceux d'émission photonique compris entre 10-9 s pour les processus autorisés par le spin, la fluorescence, et des temps beaucoup plus importants (jusqu'à plusieurs minutes ou heures) pour des processus interdits par le spin, comme la phosphorescence.
Principe
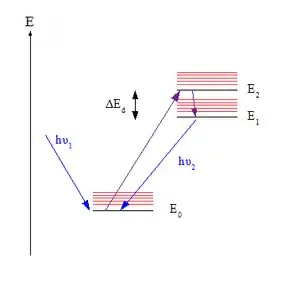






Bien que l'on puisse expliquer cette règle à partir des facteurs Franck-Condon entre transitions électroniques et l'augmentation de probabilité de ces dernières avec le recouvrement vibrationnel, ce raisonnement n'est pas correct et la loi s'applique en réalité dans tous les cas, quel que puisse être le recouvrement. Les molécules peuplées dans des états de plus grande énergie se désexcitent à grande vitesse. Entre chaque état électronique, l'énergie est désactivée (en général par dissipation dans le milieu, comme par exemple un solvant) par relaxation vibrationnelle, modifiant lors du processus la géométrie de la molécule. Durant ce procédé, des structures dans lesquelles les énergies de deux états électroniques ou plus deviennent pratiquement dégénérés d'un point de vue énergétique se produisent. En chimie quantique, ces structures sont appelées intersections coniques (IC), et traduisent des situations dans lesquelles les niveaux électroniques et vibrationnels sont couplés (l'approximation de Born-Oppenheimer n'est plus valable) et le transfert énergétique entre états devient maximale, se faisant de manière rapide, lors d'une « impulsion vibrationnelle ». Ce processus est appelé de manière générale conversion interne (s'il se produit entre états de même multiplicité de spin). La présence fréquente d'intersections coniques entre les états de plus fortes énergies propice à une désactivation très rapide depuis l'état excité le plus bas avant que toute émission radiative puisse se produire. Dans cet état excité final, la présence d'intersections coniques avec l'état inférieur, le fondamental, est moins fréquente (la différence d'énergie entre les deux états étant bien plus importante), et ainsi la molécule peut atteindre un minimum d'énergie depuis lequel elle a le temps d'émettre. La désactivation énergétique d'une molécule est toujours un équilibre entre processus radiatifs et non-radiatifs (voir rendement quantique).
Exceptions
La majorité des molécules de base en chimie possédant un état fondamental de type singulet à couche fermée, noté S0, et un état excité singulet de plus basse énergie S1, la loi de Kasha peut se formuler en indiquant que les molécules qui fluorescent le font à partir de leur état S1. Il existe des exceptions à la loi avec des cas comme celui de la fluorescence double que l'on peut observer depuis les états S1 et S2 dans l'azulène, mais de fait tous les cas de fluorescence double ne sont pas une traduction d'exception à cette règle. Cette fluorescence double peut se produire depuis deux minima distincts de la surface d'énergie potentielle de l'état S1, les deux présentant des géométries distinctes, comme dans le cas des aminobenzonitriles ou du phénylpyrrole par exemple.
Bibliographie
- Mchale, J. L. « Molecular Spectroscopy », Prentice Hall, Upper Saddle River, 1999.
- Turro, N. J. « Modern Molecular Photochemistry », University Science Books, Sausalito, 1991.
- Serrano Andrés, L.; Merchán, M., « Vida y Luz: Una Perspectiva Químico-Cuántica », Anales de Química, 100 (3), 16-31 (2004)
- Merchán, M.; Serrano Andrés, L., « Ab Initio Methods for Excited States », dans « Computational Photochemistry ». Ed. M. Olivucci, Elsevier, Amsterdam, 2004.
- Serrano Andrés, L.; Merchán, M., « Quantum chemistry of the excited state: 2005 overview », Journal of Molecular Structure: THEOCHEM 729, 99–108 (2005)
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Kasha's rule » (voir la liste des auteurs).
- (es) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en espagnol intitulé « Regla de Kasha » (voir la liste des auteurs).
 Portail de la physique
Portail de la physique  Portail de la chimie
Portail de la chimie