Chromatographie en phase liquide à haute performance
La chromatographie en phase liquide à haute performance (CLHP) — mais on trouve plus fréquemment l'abréviation anglaise HPLC (high performance liquid chromatography ou plus rarement high pressure liquid chromatography) depuis les années 1990 — est une technique de séparation analytique et/ou préparatrice de molécules présentes dans un mélange. Cela permet d'adapter les méthodes chromatographiques usuelles (voir Colonne) sur un montage haute pression.


Cette forme de chromatographie est fréquemment utilisée en biochimie, ainsi qu'en chimie analytique.
Le P du sigle, à l'origine signifiait Pression mais lorsque la méthode a été améliorée (réduction des particules et régulation de la phase stationnaire) le P a été attribué à Performance afin de marquer cette innovation.
Principe
L'échantillon à analyser est poussé par un liquide (appelé phase mobile) dans une colonne remplie d'une phase stationnaire de fine granulométrie (les « grains » sont de très petite taille). Le débit d'écoulement de la phase mobile est élevé ce qui entraîne une augmentation de la pression dans le système. Ce débit élevé diminue le temps nécessaire pour séparer les composants le long de la phase stationnaire. La fine granulométrie de la phase stationnaire permet une meilleure séparation des composants. En effet, pour un même volume de phase stationnaire la surface d'échange augmente si les « grains » qui la composent sont de diamètre plus petit. Les pics obtenus (via un détecteur à UV[1] (les protéines absorbent à 275-280 nm) relié à un système d'intégration et de calcul) sont plus étroits donc la résolution est améliorée (les pics sont bien séparés, on peut donc bien les différencier), le seuil de détection est également plus bas (des pics étroits et hauts sont plus faciles à isoler du bruit de fond que des pics larges et bas). La combinaison de ces attributs — rapidité et résolution élevées — conduit à l'appellation « haute performance».
Les phases mobiles utilisées sont des mélanges d'eau et d'un solvant organique miscible (acétonitrile, méthanol) ou des combinaisons de solvants organiques (alcools, hexane, dichlorométhane, etc.) miscibles entre eux.
Souvent, la composition de la phase mobile est modifiée au cours de l'analyse, c'est le mode dit « gradient » ou « élution graduée » (en opposition au mode « isocratique », pour lequel la composition de la phase mobile reste la même tout au long de la séparation).
Par exemple, sur une colonne apolaire, en utilisant un mélange eau/méthanol comme phase mobile, les composants les plus hydrophobes sont élués avec une concentration élevée en méthanol alors que les composants plus hydrophiles sont élués préférentiellement avec une concentration faible en méthanol. Selon la nature de la phase stationnaire et la nature des composés à séparer, on commencera par une concentration élevée en méthanol ou le contraire. L'HPLC est applicable à tous les principes chromatographiques : adsorption, partage, gel-filtration, échange d'ions, affinité, etc.
Appareillage et fonctionnement
Pompe
C'est la partie qui sert à stocker l'éluant et à l'injecter sous pression dans la colonne. Elle est composée de :
- deux pistons alternatifs ;
- réservoirs de phase mobile ;
- électrovannes ;
- amortisseur de pulsations ;
- système de purge et d'amorçage ;
- capteur de pression.
On utilise une pompe pour une élution isocratique ou plusieurs pour une élution par gradient.
Injecteur
Des tubes en acier inoxydable, en Téflon, en PEEK ou en silice fondue permettent de relier la ou les pompes à l'injecteur chromatographique. Il y a plusieurs types d'injecteurs :
- boucle d'injection : permet la répétabilité du volume d'injection ;
- injecteur seringue ;
- extraction sur phase solide en ligne.
Détecteur
Il existe plusieurs types de détecteurs :
- détecteurs spectroscopiques :
- par spectroscopie d'absorption : ultraviolet-visible (comprenant un détecteur à barrette de diodes (DAD ou PDA)) ou infrarouge,
- par spectroscopie de fluorescence ;
- réfractométrie ;
- détecteurs électrochimiques (DEC) :
- ampérométriques,
- coulométriques,
- polarographiques,
- potentiométriques ;
- conductimétrie ;
- évaporatif à diffusion de la lumière (DEDL) (evaporative light scattering detector (ELSD) en anglais) ;
- détection spectrale avec couplage :
- à la spectrométrie de masse (MS) avec la chromatographie en phase liquide-spectrométrie de masse,
- à la spectroscopie atomique.
Dégazeur
Comme son nom l'indique, ce composant permet de retirer le gaz (dioxygène) présent dans le(s) solvant(s) afin d'éviter d'endommager les échantillons ou la phase stationnaire. Deux types de dégazeurs sont utilisés en HPLC :
- dégazeur à gaz inerte : on fait barboter un gaz inerte dans la PM (Phase Mobile) pour retirer le gaz dissous dans le liquide. L'helium est le gaz inerte le plus utilisé pour cette application ;
- dégazeur par vide : cette méthode consiste à descendre en pression dans une enceinte où se trouve le solvant à l'aide d'une pompe à vide primaire, et ainsi séparer le gaz dissous dans le fluide. Elle est bien plus efficace, ne requiert plus de gaz inerte, et remplace de plus en plus l'ancienne technique dans le domaine de l'analyse HPLC. La pression dans l'enceinte est de l'ordre du millibar.
Types
Il y a plusieurs types de chromatographie en phase liquide. La nature de la phase stationnaire dépend du type de chromatographie en phase liquide que l'on veut faire ainsi que de la nature et du nombre de composés que l'on veut séparer.
Chromatographie d'adsorption
Dans cette chromatographie, la phase stationnaire consiste en une matière solide à grand pouvoir d'adsorption, telle que l'oxyde d'aluminium, les silicates de magnésium, les gels de silice. Les composants sont simplement plus ou moins retenus à la surface de la phase stationnaire par adsorption physique. C'est une technique qui prend en compte la polarité des composants.
Chromatographie de partage
Dans cette chromatographie les analytes sont séparés en fonction de leur affinité avec les phases stationnaire et mobile. L'affinité dépend de la polarité des analytes et des phases. En mode normal la phase stationnaire est polaire, en mode inverse elle est apolaire. Il y a deux types de chromatographie de partage :
- liquide - liquide : la phase stationnaire consiste en une très fine couche de liquide répartie par adsorption physique à la surface du matériau support le plus inerte possible. Les composants sont séparés comme dans une extraction liquide-liquide, sauf que la répartition des composants se fait lors du passage dans la phase liquide et non par agitation ;
- liquide - solide ou liquide - phase greffée : la phase stationnaire consiste en une espèce organique liée par des liaisons chimiques à la surface des particules du matériau support.
Chromatographie en phase normale
La chromatographie en phase normale (NPLC) consiste à séparer différents analytes selon leur polarité. Cette technique de séparation découle de l’expérience de Tswett et de son montage de chromatographie réalisé avec des pigments végétaux. Au cours des années, cette dernière a été développée davantage afin de séparer des mélanges de plus en plus complexes, en modifiant la polarité des phases et en améliorant la méthodologie de séparation analytique (ajout d’une pompe pour augmenter la pression du système et accélérer l’élution, automatisation du procédé, etc.). Celle-ci est basée sur les interactions hydrophiles et les affinités d’un analyte envers la phase mobile et stationnaire dans le but de les séparer en fonction de leur polarité[2].
Principe
La chromatographie en phase normale (NPLC) est un type de chromatographie qui fait intervenir les interactions polaires, contrairement à la chromatographie en phase inverse (RPLC), qui fait intervenir les interactions hydrophobes. Il s’agit d’un mécanisme de compétition entre la rétention d’un soluté dans la phase stationnaire et l’élution du soluté dans la phase mobile, donc des affinités de chaque analyte envers la phase mobile et la phase stationnaire. On fait donc passer un solvant non polaire ou modérément polaire dans la colonne. Les solutés les plus retenus dans la phase stationnaire seront élués en dernier alors que les solutés les moins retenus seront élués en premier. Différents types d’interactions sont exploités tels que les ponts hydrogène, les interactions dipôle-dipôle et les interactions acide-base[3].
Phases stationnaires
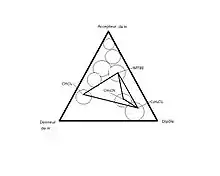
Les phases stationnaires utilisées pour une chromatographie en phase normale sont des phases polaires. Un avantage de cette chromatographie est qu’il est possible de modifier les interactions, autant celles entre les analytes et la phase stationnaires, que celles avec les analytes et la phase mobile et la phase mobile avec la phase stationnaire. Puisque l’analyte se déplace en fonction de son affinité pour la phase mobile et la phase stationnaire, il est possible de choisir les deux phases afin d’optimiser la séparation d’un système complexe. On peut directement utiliser la silice puisqu’elle contient des groupements silanols (Si-OH) en surface. Selon la méthode utilisée pour la synthèse de la silice, la surface spécifique des particules, le diamètre des particules ainsi que le diamètre des pores varie grandement. Chaque type de silice possède ses propres propriétés de séparation. Des types des silices contenant plus de groupement silanols libres (plus acide) ou moins de groupements silanols peuvent aussi être utilisés en fonction du type de séparation désiré. Il est aussi possible de fonctionnaliser la silice afin de faire varier la polarité ou le caractère de la silice en surface, tout en rendant sa surface plus homogène. Selon le type de séparation que l’on désire faire, on doit choisir entre trois classes de silice fonctionnalisée suivant le triangle de sélectivité suivant :
- Accepteur de proton ;
- Donneur de proton ;
- Interactions dipôle-dipôle.
Si la séparation ne fonctionne pas avec un type de silice, on choisit un second type qui se situe dans une autre classe afin d’engendrer le changement le plus drastique des interactions entre la phase stationnaire et les analytes. Les silices fonctionnalisées les plus souvent utilisées pour la chromatographie en phase normale sont la silice cyanopropylée, la silice aminopropylée et la silice 1,2-hydroxypropylée (diol). D’autres groupements peuvent aussi être utilisés pour la fonctionnalisation de la silice pour des applications plus spécifiques, mais ceux-ci suffisent en général. La colonne diol est une colonne acide, la colonne amino est une colonne plutôt basique alors que la colonne cyano fait des interactions dipôle dipôle modérées.
Phase mobile
Les solvants utilisés sont les mêmes que ceux utilisés pour les autres types de chromatographie. Idéalement, on utilise des solvants peu visqueux, afin de permettre un débit d’éluant plus élevé dans la colonne, dont le point d’ébullition est bas pour faciliter la récupération des analytes après élution. De plus, pour faire la chromatographie, le solvant doit absolument être moins polaire que les analytes pour éviter une élution directe des analytes. En général, on utilise l’hexane ou le pentane comme solvant non polaire de base (que l’on appelle le solvant A). Il est possible de jouer sur la polarité de l’éluant en faisant varier la polarité du solvant A. Il est par contre plus simple d’utiliser un éluant binaire puisque la polarité variera en fonction de la quantité d’un solvant polaire ajouté. Les solvants polaire, aussi appelé solvants modificateurs (solvant B), communément utilisés sont le chloroforme, le dichlorométhane, l’acétate d’éthyle, le tétrahydrofurane, le méthyl tert-butyl éther (MTBE), l’éther diéthylique ainsi que des alcools comme le méthanol ou l’isopropanol.
Un autre facteur important concernant la phase mobile est la force du solvant. Puisque la technique est basée sur les différentes interactions entre les analytes, la phase mobile et la phase stationnaire, il est donc important d’ajuster la force de l’éluant, que l’on définit par l’affinité de la phase mobile pour la phase stationnaire. Ainsi, un solvant dont la force d’élution est supérieure à une autre aura plus de facilité à entrainer un analyte dans la phase mobile, puisque ses interactions seront plus fortes avec la phase stationnaire. Aucune échelle absolue de force de solvants n’existe puisque celle-ci varie d’une colonne à l’autre. Par contre, plusieurs échelles empiriques permettent de classer les solvants dans un ordre correspondant à leur force de solvant (ɛ0). Pour un mélange de solvant, la force de solvant n’augmente pas linéairement avec le pourcentage de solvant polaire ajouté. Il peut par contre être calculé à l’aide de l’équation suivante :

Où εAB est la force du mélange de solvant, εA et εB la force de solvant de chacun des solvants purs A et B respectivement, NB la fraction molaire et nB la surface moléculaire du solvant modificateur et α le coefficient d’activité, habituellement 1 pour une colonne analytique moderne. Lorsqu’on veut optimiser une séparation, il est important de conserver la même force de solvant après changement de solvant B.
Sélectivité du solvant
La sélectivité du solvant est l’ensemble des interactions acide-base, dipôle-dipôle et liaisons hydrogène entre les molécules de solvant et l’analyte. Il y a un changement dans la sélectivité du solvant quand les interactions moléculaires entre la phase stationnaire, le soluté et la phase mobile changent brusquement avec le changement de solvant. La sélectivité du solvant est basée sur le triangle de sélectivité de Snyder, qui guide le choix du solvant ou des solvants à choisir ainsi que la phase stationnaire pour optimiser la séparation de l’analyte. Chaque apex du triangle représente une caractéristique d’un solvant, soit accepteur de proton, donneur de proton ou ayant un dipôle. Les solvants sont classés dans le triangle, de façon à les catégoriser selon les interactions possibles du solvant avec l’analyte. Pour changer subitement les interactions entre le solvant et les analytes, il suffit de passer d’un solvant près d’un apex du triangle vers un solvant qui se situe plus près d’un autre apex[2],[4].
Effet de délocalisation (déplacement)
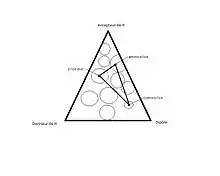
L’effet de localisation est un effet non négligeable en NPLC. Lorsqu’un soluté polaire peut faire des interactions fortes (dipôle ou pont hydrogène) avec les groupements fonctionnels en surface de la silice, on dit que le soluté se « localise ». En effet, un soluté a une capacité de localisation qui est donnée en fonction de la surface efficace du groupement en surface de la silice. Cette surface peut être réduite par l’action du solvant modificateur, qui exerce, en compétition avec le soluté, des interactions fortes du même type. Plus le solvant a d’interactions avec les groupements en surface, plus la rétention du soluté est diminuée. Le solvant peut donc éluer plus rapidement un soluté dans la colonne si ses interactions avec la phase stationnaire sont suffisantes pour contrer celles du soluté; on dit qu’il a un effet de délocalisation. Parfois, l’effet de délocalisation est prédominant par rapport à l’acidité, la basicité ou la dipolarité d’un solvant. Il a donc un effet majeur sur la rétention des solutés.
Effets secondaires de solvant
Un autre type d’effets concernent les interactions entre le solvant et le soluté. On les appelle les effets secondaires de solvant. Ces effets peuvent, dans certains cas, améliorer la rétention des solutés, dans d’autres, y nuire. Un premier type d’effet secondaire de solvant est lorsque le solvant interagit directement avec le soluté. La force de rétention est donc répartie et la phase stationnaire a de la difficulté à contrer ces effets, ce qui engendre une diminution de la rétention. Un deuxième effet secondaire se produit lorsque la concentration du solvant modificateur est supérieure dans la phase stationnaire que dans la phase mobile. Lorsque le soluté fait des interactions fortes avec le solvant modificateur, cet effet est bénéfique et augmente la rétention du soluté. Un troisième effet secondaire consiste en une force si grande entre le solvant modificateur et la phase stationnaire que le solvant modificateur est stériquement entravé, ce qui diminue la rétention étant donné que le solvant modificateur dans la phase mobile peut toujours faire des interactions avec le soluté.
Chromatographie en phase inverse
La base d'une phase inverse est une phase normale sur laquelle des chaînes alkyles (ou autres selon la polarité recherchée) ont été greffées au niveau des groupes silanols (end-capping).
En général, la phase stationnaire est majoritairement composée de petites particules de silice sur lesquelles on a greffé des fonctions chimiques, le plus souvent de chaines alkyle à 8 ou 18 atomes de carbone.
Les fonctions silanols (Si-OH) qui subsistent engendrent des interactions hydrophiles parasites, qui rendent les résultats non reproductibles surtout pour les molécules basiques. Pour éviter cela, la surface de la silice est généralement recouverte par une fonction méthyle et les fonctions silanols ne sont plus libres mais sous la forme (Si-O-CH3), c'est cette étape que l'on appelle « end-capping ». Les fonctions chimiques utilisées pour le end-capping peuvent toutefois être de nature très diverses et les colonnes de dernières générations résistant à des pH extrêmes sont généralement end-capped avec des fonctions proposant une plus grande gène stérique, telles que le tert-butyle (Si-O-C(CH3)3).
Selon le taux de greffage, on obtient une plus ou moins grande résolution.
Cette phase stationnaire est dite « inverse » car de polaire et hydrophile (sans les « greffes »), la phase devient apolaire et hydrophobe.
Chromatographie par échange d'ions
La phase solide est une résine insoluble munie de groupes fonctionnels capable de dissocier. Ce sont, habituellement, des groupes « acide sulfonique » (SO3H) pour les échangeurs de cations et « ammonium quaternaire » (N(R)3) pour les échangeurs d'anions.
Chromatographie d'exclusion stérique
Les composants sont séparés selon leur dimension moléculaire. La phase stationnaire est composée d'un matériau poreux (petites particules de silice ou de polymères), les molécules dont le diamètre est supérieur à celui des pores ne peuvent pénétrer et ne sont pas retenues. La durée de séjour dans la colonne augmente lorsque la taille des analytes diminue.
Chromatographie chirale
Cette technique de chromatographie consiste en la formation de liaisons non covalentes entre les énantiomères du substrat et l'absorbant chromatographique chiral donnant des complexes diastéréoisomères ayant des affinités de liaisons différentes. Elle sert donc en particulier à séparer des énantiomères.
Paramètres de chromatographie
La qualité et la durée d'une séparation peuvent changer avec la nature de la phase stationnaire et de la phase mobile utilisées. Mais pour un même type de phase stationnaire et une même phase mobile, la qualité et la durée d'une séparation peuvent aussi être modifiées par les caractéristiques géométriques et opératoires de ces deux phases.
Caractéristiques géométriques de la phase stationnaire
- La longueur L et le diamètre interne dc de la colonne.
- Le diamètre des particules de phase stationnaire dp.
Caractéristiques opératoires de la phase mobile
- La vitesse linéaire de l'écoulement u.
- La perte de charge ou pression appliquée ΔP entre l'entrée et la sortie de la colonne (la pression de sortie étant généralement atmosphérique). Elle est donnée par la loi de Darcy

où est la viscosité de la phase mobile et Φ est le facteur de résistance à l'écoulement qui dépend de la forme des particules et de la qualité du remplissage de la phase stationnaire.
Pour des colonnes bien remplies de particules sphériques ou irrégulières, la formule empirique de Vérillon[5]


établie avec les unités usuelles, permet le calcul commode d'une estimation de ΔP en fonction du débit F de la phase mobile avec une incertitude inférieure à 25 %, et dans un large domaine de conditions opératoires (2 μm < dp < 50 μm, 1 mm < dc < 100 mm) pour les chromatographies en phases liquide et supercritique.
Remarque : la loi de Darcy est à l'hydraulique ce que la loi d'Ohm est à l'électricité. La pression hydraulique est analogue au potentiel électrique, le débit de liquide est analogue à l'intensité du courant, la perméabilité hydraulique est analogue à la conductivité électrique, la pressurisation et la dépressurisation d'une colonne chromatographique sont respectivement analogues à la charge et à la décharge d'un condensateur électrique.
Grandeurs caractéristiques en chromatographie
Le résultat observable d'une analyse HPLC se présente sous la forme d'une courbe du signal détecté en fonction du temps : c'est le chromatogramme. Il comporte plusieurs pics de forme gaussienne, de caractéristiques différentes :
- le temps de rétention ou tR, temps du maximum du pic. On appelle t0 ou temps de rétention nulle le temps correspondant à un composé non retenu chromatographiquement ;
- la largeur du pic, mesurée à mi-hauteur : ω1/2, ou à sa base, par l'intersection des tangentes du pic à ses points d'inflexion avec la ligne de base : ω.
De là peuvent être calculées plusieurs caractéristiques de la colonne pour la séparation des pics :
- le facteur de rétention k' du pic, par la formule

- l'efficacité N ou nombre de plateaux théoriques, reliée à la largeur du pic par la formule
- Cette valeur mesure la finesse du pic. À partir de cette valeur peut être calculée la hauteur équivalente à un plateau théorique (HEPT) H, qui permet de comparer des colonnes de longueur différente


- la sélectivité α entre deux pics 1 et 2 (2 étant plus retenu que 1), définie par le rapport de leurs deux facteurs de rétention
- Elle mesure la capacité de la colonne à séparer les maxima des pics. Plus elle est supérieure à 1, plus les temps de rétention sont éloignés ;

- la résolution RS entre les pics 1 et 2
- Cette valeur mesure la qualité de séparation et d'absence de recouvrement entre les deux pics considérés.
- La résolution ainsi définie est l'objectif de la séparation chromatographique. C'est une fonction des trois caractéristiques (efficacité, sélectivité et rétention), elles-mêmes définies ci-dessus, dont l'expression est
- Cette expression montre notamment qu'une même résolution peut être obtenue dans des conditions chromatographiques ou bien très efficaces (optimisation cinétique) ou bien très sélectives (optimisation thermodynamique).


Quoi qu'il en soit, la performance qui caractérise la technique HPLC est définie par le pouvoir de résolution par unité de temps, ce qui n'implique pas systématiquement l'utilisation de la pression la plus haute possible.
Notes et références
- Résultat de l'échantillonnage des cannabinoïdes d'une huile de CBD avec la Chromatographie liquide haute performance (HPLC) couplée à la détection UV - Naturicious.
- (en) William T. Cooper, Normal Phase Liquid Chromatography, Encyclopedia of Analytical Chemistry, .
- (en) Snyder, L.R. et Kirkland, J.J., Introduction to the Modern Chromatography, John Wiley & Sons, , 2e éd.
- (en) Johnson, Andrew R. et Vitha, Mark F., « Chromatographic selectivity triangles », Journal of Chromatography A, vol. 1218, n.4, , p.559-560.
- J. L. Cuq, 2007, La Chromatographie Liquide, université Montpellier 2, p. 13-14.
Articles connexes
- UPLC (Ultra Performance Liquid Chromatography)
- Liste de sigles de biologie cellulaire et moléculaire
- Techniques de biologie moléculaire
 Portail de la chimie
Portail de la chimie  Portail de la biochimie
Portail de la biochimie