Myopathie de Bethlem
La myopathie de Bethlem est une maladie génétique musculaire de transmission autosomique dominante en rapport avec des mutations des gènes Col6A1, Col6A2 et Col6A3 codant le collagène type VI. Elle affecte particulièrement les muscles du squelette, utilisés pour le mouvement[1].
| OMIM | 158810 |
|---|---|
| DiseasesDB | 32019 |
![]()
Caractéristiques
La myopathie de Bethlem, aussi appelée dystrophie musculaire[2] congénitale bénigne, syndrome de Leonard ou myopathie congénitale bénigne à contractures[1], est une forme bénigne de myopathie lentement progressive et une collagénopathie. Comme toutes les myopathies, il s'agit d'une dégénérescence du tissu musculaire[3]. C'est une maladie extrêmement rare, moins de 100 cas ont été rapportés dans la littérature, soit une prévalence estimée entre un cas sur 200 000 et un cas sur plus d'un million. Moins de trente cas sont recensés en France[4]. Des mutations sur l'une des trois sous-unités, Col6A1, Col6A2 et Col6A3[5], les deux premiers sur le chromosome 21, le troisième sur le chromosome 2[4], et codant le collagène[2] VI, sont à l'origine de la maladie. Le collagène VI est alors anormal ou produit en faible quantité, ce qui entraîne une rupture de la matrice extracellulaire entourant les cellules musculaires[1].
La taille et le schéma d'expression des gènes constituent, cependant, des freins aux études moléculaires[6]. D'abord décrites comme deux maladies distinctes, la dystrophie congénitale musculaire d'Ullrich et la myopathie de Bethlem sont des manifestations différentes (phénotypes) de la même maladie. La distinction clinique est difficile à la naissance, mais, au moment de l'acquisition de la marche, le diagnostic de la myopathie de Bethlem est assuré. Également, on n'observe pas de contractures dans la myopathie d'Ullrich. La distinction est importante, car ces deux maladies ont des modes de transmissions différentes.
Manifestations cliniques
Les manifestations cliniques de la myopathie de Bethlem diffèrent peu de celles observées dans les autres formes légères de dystrophie musculaire, à l'exception de rétractions des doigts[6]. La myopathie de Bethlem est caractérisée par la combinaison de faiblesse musculaire et de contractures musculaires atteignant principalement les muscles des doigts, du poignet, du coude et de la cheville[1]. Le début peut apparaître à tout âge. Prénatal, il s'accompagne d'une diminution des mouvements fœtaux, néonatal, d'hypotonie et de torticolis provoquant l'inclinaison latérale de la tête[1].
Dans l'enfance, l'apparition de la maladie s'accompagne de faiblesse musculaire, d'hypotonie (torpeur), de pied-bot, de torticolis (raideur de la nuque)[7] et de contractures, ainsi que d'un retard à l'acquisition de la marche et de la position assise[1]. Le signe de Gowers, marqué par l'ascension des cuisses à l'aide des mains, lorsque l'enfant se relève du sol, et la marche sur la pointe des pieds, due aux contractures, est également caractéristique.
À l'âge adulte, entre 40 et 60 ans, on observe des faiblesses musculaires, une faible endurance, des difficultés à monter les escaliers, et des contractures des muscles fléchisseurs des doigts et des muscles des jambes, notamment des raideurs des tendons d'Achille. Les malades ont du mal à lever les bras au-dessus de leur tête[7]. Certains patients présentent des anomalies de la peau. Ce sont, par exemple, de petites bosses, appelées hyperkératoses folliculaires, qui se développent autour des coudes et des genoux. C'est aussi, parfois, une peau douce et velouteuse dans les paumes et sur la plante des pieds, ou bien des blessures qui s'ouvrent, accompagnées des petits saignements, et se creusent au cours du temps, créant des cicatrices peu profondes[1]. La peau des bras et des jambes peut aussi devenir sèche ou rugueuse (le toucher est décrit comme analogue à celui d'un poulet plumé)[7].
À cause de cette pathologie, plus des deux tiers des personnes de plus de 50 ans atteintes par cette maladie ont besoin d'une aide (canne[7], déambulateur ou chaise roulante[1]) pour les déplacements extérieurs. L'insuffisance musculaire implique rarement les muscles respiratoires et le diaphragme. Aucun effet sur le muscle cardiaque n'a été observé[7]. Il semble également que les muscles de l'utérus ne soient pas affectés, autorisant des accouchements normaux[8].
Diagnostic
Le taux de créatine kinase dans le sérum, qui est élevé, à cause de la mort des cellules musculaires, et les observations histologiques, réalisées au microscope[7], ne permettent pas d'établir le diagnostic à eux seuls[6]. Le diagnostic est établi par une étude, par immunohistochimie, de la synthèse et de la sécrétion du collagène 6, sur des fibroblastes obtenus après mise en culture d’une biopsie de peau. Le taux de laminine β 1, une protéine caractéristique, est également mesuré, sa faiblesse étant un indicateur de la maladie[7]. Les analyses génétiques sont réalisées, dans un second temps, par séquençage des Acides ribo-nucléiques (ARN) messagers extraits de ces fibroblastes[9]. Comme la maladie est extrêmement rare, les résultats peuvent attendre des mois, voire des années[7].
Diagnostic différentiel
- Maladie de Kugelberg-Weilander[10].
- La dystrophie musculaire d'Emery-Dreifuss (EDMD) présente, comme la myopathie de Bethlem, les contractures des coudes et des chevilles. Cependant, celles du coude et de la colonne vertébrale sont beaucoup plus sévères dans le cas de l'EDMD. La détection de défauts de conduction cardiaque permet d'assurer le diagnostic[4].
- Certains sous-types (1A à 1G) de la dystrophie musculaire des membres inférieurs (LGMD) autosomique dominante peuvent être différenciés, grâce à l'identification des gènes mis en cause[4].
- Formes atypiques de la dystrophie musculaire congénitale sporadique[4].
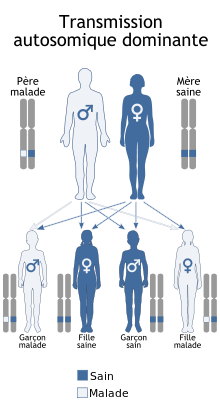
Transmission
La myopathie de Bethlem se transmet de façon autosomique dominante[2], ce qui signifie qu'une seule copie par cellule du gène altéré est suffisante. La plupart des cas résulte d'une nouvelle mutation du gène, chez des patients n'ayant pas d'antécédents familiaux. Dans quelques cas, la maladie est transmise par un parent affecté. Dans de rares cas, la transmission est autosomique récessive et il faut deux copies par cellule du gène pour que la maladie s'exprime. Dans ce cas, les deux parents portent une copie du gène altéré, mais ne présentent généralement pas les symptômes de la maladie[1].
Traitement
Le traitement de la myopathie de Bethlem est purement palliatif[6]. Il fait appel à la kinésithérapie. Les physiothérapeutes proposent des exercices permettant d'assouplir les tendons contractés et de conserver la flexibilité des muscles[11]. Des opérations du tendon d'Achille permettent de diminuer les contractures et cette chirurgie peut être répétée plusieurs fois entre l'enfance et l'âge adulte[2]. Par ailleurs, un régime est souvent prescrit, afin d'éviter la surcharge pondérale, qui est un facteur aggravant des difficultés musculaires[11].
Complications
La myopathie de Bethlem peut entraîner des affections pulmonaires, si la toux n'est pas assez forte, à cause de la faiblesse des muscles respiratoires[7]. Des tests respiratoires doivent être pratiqués régulièrement. La vaccination contre la grippe est fortement recommandée et les maladies pulmonaires doivent être rapidement traitées par antibiothérapie[11].
Des problèmes de constipation apparaissent parfois. Ils sont traités par un régime riche en fibres. Il est recommandé de boire beaucoup. Les laxatifs sont très rarement employés[11].
Recherche
Un groupe de recherche, comprenant des chercheurs et des cliniciens et financé par l'Union européenne, Myo-Cluster, recense les cas connus en Europe et les mutations des gènes mis en cause[4].
En 2003, les professeurs Paolo Bernardi et Paolo Bonaldo, de l'Université de Padoue, ont créé une souris présentant le même type d'affection que dans les myopathies de Bethlem et d'Ullrich, en éliminant le gène Col6A1. Grâce à ce modèle, ils ont pu proposer un mécanisme expliquant pourquoi l'absence de cette protéine entraîne la mort des cellules musculaires. Ils ont pu également identifier une molécule, la CsA, capable de bloquer ce processus de dégénérescence cellulaire. En janvier 2007, la même équipe synthétise un analogue de la CsA, la Debio 025, qui présente la même efficacité sur le muscle sans affecter le système immunitaire[12].
Classification
- Numéro Orphanet : ORPHA610.
- Code CIM 10 : G71.0.
Histoire
La myopathie de Bethlem est découverte, en 1976, par les médecins néerlandais J. Bethlem[7] et Wijngaarden[4].
Notes
- Bethlem myopathy - Genetics Home Reference .
- G. J. Jobsis, J. M. Boers, P. G. Barth, M. de Visser, « Bethlem myopathy: a slowly-progressive congenital muscular dystrophy with contractures », dans Brain, vol. 122, n° 4, 1999, p. 649 à 655.
- Myopathie : Symptômes - Causes - Diagnostics - Traitements .
- Bethlem myopathy .
- A. K. Lampe, K. M. Bushby, « Collagen VI related muscle disorders », dans J. Med. Genet., vol. 42, n° 9, septembre 2005, p. 673 à 685.
- Orphanet: Myopathie de Bethlem .
- Newcastle Hospitals - Bethlehem Myopathy .
- Bethlem Myopathy - Neuromuscular Diseases Message Board - HealthBoards .
- Collagénopathies (Myopathies d'Ullrich et de Bethlem) .
- Bethlem Myopathy > Home .
- Bethlem myopathy | Muscular Dystrophy Campaign .
- Rencontre avec le Pr Bernardi .
Références
- (fr) J. Bethlem, Myopathies, éd. North-Holland Pub. Co., 1977.
- (fr) G. Serratrice, Les contractures musculaires, éd. Presse Méd, 1999.
- (fr) Nathalie Vincent-Lacaze, « Thérapie génique des dystrophies musculaires », dans Annales de l’Institut Pasteur/Actualités, vol. 10, n° 3-4, juillet-décembre 1999, p. 327 à 338.
- (fr) J. A. Lobrinus, « Apport de la pathologie dans les maladies neuromusculaires », dans Revue médicale suisse, n° 2460, 2003.
- (fr) Florence Ruggiero, Muriel Roulet, Christelle Bonod-Bidaud, « Les collagènes du derme : au-delà de leurs propriétés structurales », dans Journal de la Société de Biologie, vol. 199, n° 4, 2005, p. 301 à 311.
- (fr) « Myopathie de Bethlem », dans Myoline, vol. 96, 2008, p. 4.
- (en) M. D. Mohire, R. Tandan, T. J. Fries, B. W. Little, W. W. Pendlebury, W. G. Bradley, « Early-onset benign autosomal dominant limb-girdle myopathy with contractures (Bethlem myopathy) », dans Neurology, vol. 38, 1988, p. 573 à 580.
- (en) M. Fardeau, « Congenital myopathies », dans Myology, 1994.
- (en) L. Merlini, L. Morandi, C. Granata, A. Ballestrazzi, « Bethlem myopathy: early-onset benign autosomal dominant myopathy with contractures: description of two new families », dans Neuromusc. Disord., vol. 4, 1994, p. 503 à 511.
- (en) G. J. Jöbsis, P. A. Bolhuis, J. M. Boers, F. Baas, R. A. Wolterman, G. W. Hensels, M. de Visser, « Genetic localization of Bethlem myopathy », in Neurology, vol. 46, 1996, p. 779 à 782.
- (en) G. J. Jöbsis, H. Keizers, J. P. Vreijling, M. de Visser, M. C. Speer, R. A. Wolterman, F. Baas, P. A. Bolhuis, « Type VI collagen mutations in Bethlem myopathy, an autosomal dominant myopathy with contractures », dans Nat. Genet., vol. 14, 1996, p. 113 à 115.
- (en) M. C. Speer, R. Tandan, P. N. Rao, T. Fries, J. M. Stajich, P. A. Bolhuis, G. J. Jöbsis, J. M. Vance, K. D. Viles, K. Sheffield, C. James, S. G. Kahler, M. Pettenati, J. R. Gilbert, P. H. Denton, L. H. Yamaoka, M. A. Pericak-Vance, « Evidence for locus heterogeneity in the Bethlem myopathy and linkage to 2q37 », dans Hum. Mol. Genet., vol. 5, 1996, p. 1 043 à 1 046.
- (en) G. J. Jöbsis, J. M. Boers, P. G. Barth, M. de Visser, « Bethlem myopathy: a slowly progressive congenital muscular dystrophy with contractures », dans Brain, vol. 122, n° 4, 1999, p. 649 à 655.
- (en) G. Pepe, S. Lucioli, O. Camacho Vanegas, C. Minosse, B. Giusti, J. A. Urtizberea, F. Muntoni, K. Bushby, M. de Visser, C. Bönnemann, P. Sabatelli, E. Bertini, L. Merlini, M. L. Chu, « Genotype-phenotype correlation in Bethlem myopathy », dans Neuromusc. Disord., vol. 12, 2002, p. 720.
- (en) A. J. van der Kooi, W. G. de Voogt, E. Bertini, K. Bushby, L. Merlini, F. Muntoni, B. Talim, G. A. Urtizberea, M. de Visser, « Cardiac and pulmonary investigations in Bethlem myopathy », dans Neuromuscul. Disord., vol. 12, 2002, p. 723.
- (en) « Novel mutations in collagen VI genes: Expansion of the Bethlem myopathy phenotype », dans Neurology, vol. 58, n° 4, 2002, p. 593 à 602.
- (en) « Kinked Collagen VI Tetramers and Reduced Microfibril Formation as a Result of Bethlem Myopathy and Introduced Triple Helical Glycine Mutations », dans J. Biol. Chem., vol. 277, n° 3, 2002, p. 1 949 à 1 956.
- (en) « Detection of common and private mutations in the COL6A1 gene of patients with Bethlem myopathy », dans Neurology, vol. 64, n° 11, 2005, p. 1 931 à 1 937.
- (en) « Automated genomic sequence analysis of the three collagen VI genes: applications to Ullrich congenital muscular dystrophy and Bethlem myopathy », dans J. Med. Genet., vol. 42, n° 2, 2005, p. 108 à 120.
- (en) « Cardiac and Pulmonary Investigations in Bethlem Myopathy », dans Arch Neurol, vol. 63, n° 11, 2006, p. 1 617 à 1 621.
- (en) D. Hicks, A. K. Lampe, R. Barresi, R. Charlton, C. Fiorillo, C. G. Bonnemann, J. Hudson, R. Sutton, H. Lochmüller, V. Straub, K. Bushby, « A refined diagnostic algorithm for Bethlem myopathy », dans Neurology, vol. 10, n° 14, 2008, p. 1 192 à 1 199.
- (en) « Autosomal recessive Bethlem myopathy », dans Neurology, vol. 73, n° 22, 2009, p. 1 883 à 1 891.
Voir aussi
- Chromosome 21 humain.
- Dystrophie musculaire des ceintures.
- Maladies en rapport avec le collagène type VI.
- Maladies héréditaires du collagène.
- Myopathie.
Lien externe
