Gène et protéine CFTR
Dans l'espèce humaine, le gène CFTR (pour Cystic fibrosis transmembrane conductance regulator) code une protéine CFTR membranaire responsable du flux d'ions chlorure. La protéine forme un canal perméable aux ions chlorure et thiocyanate[1], des cellules épithéliales. Chez l'humain, le gène CFTR est localisé sur le locus 7q31.2, donc dans la sous-bande 2 de la bande 1 de la region 3 du bras long du chromosome 7. Il est constitué d'environ 250 000 paires de bases et contient 27 exons[2],[3]. Des mutations de ce gène sont responsables de la mucoviscidose, la maladie génétique la plus fréquente en Europe, et de l'aplasie congénitale du canal déférent (CAVD).

Histoire
L'étude de la transmission de la mucoviscidose dans de grandes familles aboutit à la découverte en 1985 de liaisons génétiques entre la maladie et deux marqueurs génétiques:
- le gène PON qui code une enzyme, la paraxonase plasmatique.
- le fragment RFLP appelé DOCRI 917
Le gène CFTR fut découvert en 1989 par une équipe canadienne.
La découverte de ce gène est un bon exemple de génétique inverse : on a d'abord localisé la région où se trouvait le gène, pour ensuite découvrir sa séquence, et ce n'est qu'ensuite qu'on a pu déterminer quel était le rôle de ce gène (en l'occurrence, un rôle dans l'échange d'ions Cl- entre cellule et environnement.)
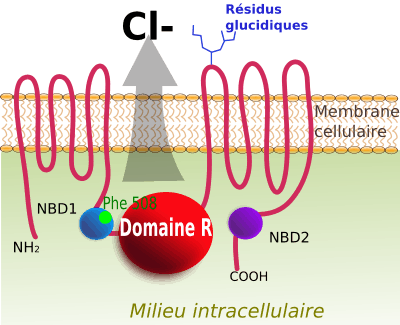
Rôles
La protéine est exprimée dès la vie fœtale[4].
Il s'agit d'un canal ionique régulé par l'AMP cyclique[5].
La protéine CFTR est impliquée dans le non transport des halogènes Chlore (Cl-), Iode (I-), Brome (Br-) et pseudohalogene Thiocyanate (SCN-) chez les personnes atteintes de mucoviscidose, entraînant des pertes de sueur riches en sodium. De fait, les composés du système endogène de défense immunitaire[6] du poumon de type hypoiodite (OI-), hypochlorite (OCl-), hypothiocyanite (OSCN-) ne peuvent être produits et de fait, les mécanismes de défenses antimicrobiennes sont inhibés[7],[8].
Elle permet également l'hydratation du mucus en favorisant la sécrétion d'eau et en inhibant une pompe à sodium épithéliale[9]. Elle favorise aussi le passage d'ions bicarbonates dans le mucus et contrôle ainsi son acidité[10]. Cette alcalinisation pourrait être importante car un mucus trop acide a des propriétés antibactériennes moindres[11].

Cible thérapeutique
L'ivacaftor permet d'augmenter la probabilité de l'ouverture d'un canal CFTR[12]. La molécule semble pallier certains symptômes de la mucoviscidose par ce biais[13].
Notes et références
- (en) M. Childers, G. Eckel, A. Himmel et J. Caldwell, « A new model of cystic fibrosis pathology: lack of transport of glutathione and its thiocyanate conjugates. », Med Hypotheses, vol. 68(1), , p. 101-12 (lire en ligne)
- (en) « CFTR: The Gene Associated with Cystic Fibrosis », sur http://www.ornl.gov, Genomics.energy.gov, (consulté le 6 juin 2008)
- (en) « CFTR », sur http://ghr.nlm.nih.gov, Genetics Home Reference, (consulté le 6 juin 2008)
- (en) Ann E.O. Trezise, Julie A. Chambers, Catherine J. Wardle, Stephen Gould et Ann Harris, « Expression of the cystic fibrosis gene in human foetal tissues », Human Molecular Genetics, vol. 2, no 3, , p. 213-218 (lire en ligne)
- (en) Stutts MJ, Canessa CM, Olsen J, « CCFTR as a cAMP-dependent regulator of sodium channels » Science, 1995;269:847-850
- (en) Gregory E. Conner, Corinne Wijkstrom-Frei, Scott H. Randell, Vania E. Fernandez et Matthias Salathe, « The Lactoperoxidase System Links Anion Transport To Host Defense in Cystic Fibrosis », FEBS Lett., vol. 581, no 2, , p. 271-278 (PMID 17204267, PMCID PMC1851694, lire en ligne)
- (en) Fischer H. « Mechanism and function of DUOX in epithelia of the lung » Antioxid Redox Signal. 2009;11(10):1-13. https://www.ncbi.nlm.nih.gov/pubmed/19358684
- (en) Xu Y, Szep S, Lu Z. « The antioxidant role of thiocyanate in the pathogenesis of cystic fibrosis and other inflammation related diseases », PNAS. 2009; Early edition, November 16th http://www.pnas.org/content/106/48/20515.full.pdf+html
- (en) Boucher RC, « Airway surface dehydration in cystic fibrosis: pathogenesis and therapy » Annu Rev Med, 2007;58:157-170
- Poulsen JH, Fischer H, Illek B, Machen TE, Bicarbonate conductance and pH regulatory capability of cystic fibrosis transmembrane conductance regulator, Proc Natl Acad Sci U S A, 1994;91:5340-5344
- Stoltz DA, Meyerholz DK, Welsh MJ, Origins of cystic fibrosis lung disease, N Engl J Med, 2015;372:351-362
- Van Goor F, Hadida S, Grootenhuis PD et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770, Proc Natl Acad Sci U S A, 2009;106:18825-18830
- Ramsey BW, Davies J, Elvaney G et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation, N Engl J Med, 2011;365:1663-1672
