Anomalie d'Ebstein
L'anomalie d'Ebstein, également dénommée malformation d'Ebstein est une cardiopathie congénitale rare caractérisée par un défaut de la formation de la valve tricuspide qui sépare l'oreillette droite du ventricule droit.
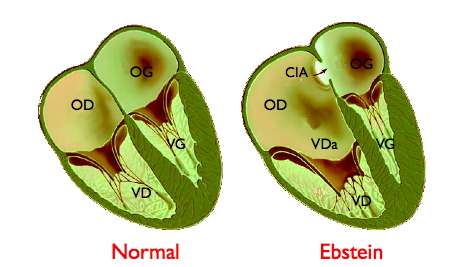
Description
Normalement, la valve tricuspide est constituée de trois feuillets (septal, antérieur et postérieur), insérés sur l'anneau auriculo-ventriculaire et s'ouvrant sensiblement dans le même plan que la valve mitrale, son homologue située entre l'oreillette gauche et le ventricule gauche.
Dans la malformation d'Ebstein, l'ouverture des feuillets septal et postérieur est déplacée vers la pointe du ventricule droit, à une distance plus ou moins grande de l'anneau auriculo-ventriculaire et il s'associe habituellement un certain degré de malformation du 3° feuillet antérieur. Cette anomalie résulte d'un défaut de clivage de la valve au cours de la vie fœtale. Pour plus de détail, consulter ci-dessous le paragraphe consacré à l'embryologie de cette malformation.
En plus de cette anomalie d'implantation, la valve tricuspide malformée est le siège d'une fuite (insuffisance tricuspide) quasiment constante mais d'intensité variable. Dans 1/4 des cas, il s'y associe un rétrécissement valvulaire.
Cette anomalie est responsable d'une modification de l'anatomie fonctionnelle du cœur droit. Normalement, l'oreillette droite (OD) et le ventricule droit (VD) se contractent successivement, la contraction de l'oreillette se produisant alors que la valve tricuspide est ouverte, celle du ventricule droit après qu'elle s'est fermée, pour éviter un reflux de sang dans l'oreillette. L'implantation trop basse de la valve tricuspide modifie la répartition des cavités. L'oreillette droite est dilatée, formée de l'oreillette normale et de la partie proximale du ventricule droit (portion « auricularisée » - VDa sur le schéma). Cette oreillette a une cinétique anormale, avec une double contraction se produisant aux temps auriculaire et ventriculaire du cycle cardiaque. Le ventricule droit est quant à lui de dimensions réduites, au prorata de l'importance du déplacement de la valve tricuspide.
Malformations cardiaques associées
Dans un tiers à la moitié des cas, d'autres malformations cardiaques lui sont associées:
- Les plus fréquentes sont :
- La présence d'une communication inter-auriculaire (CIA sur le schéma) ou d'un foramen ovale perméable (voir Circulation fœtale), observée dans 50 % des cas au moins, (90 % pour certains auteurs) et qui modifie profondément la symptomatologie en transformant l'anomalie d'Ebstein en cardiopathie cyanogène par passage anormal de sang de l'oreillette droite dans l'oreillette gauche.
- La persistance de voies de conduction électrophysiologiques anormales entre les oreillettes et les ventricules, qualifiées également de "voies accessoires", susceptibles de se traduire par un Syndrome de Wolff-Parkinson-White sur l'ECG et de provoquer des troubles du rythme cardiaque (25 % des cas au moins).
- Une sténose pulmonaire, qui aggrave considérablement le pronostic.
Dans les associations plus rares, signalons celle d'une anomalie d'Ebstein et d'une transposition corrigée des gros vaisseaux, deux malformations pourtant exceptionnelles.
Fréquence et facteurs favorisants
Cette malformation est rare, de l'ordre de 1 à 2 pour 100 000 naissances. Elle représenterait environ 0,5 % de l'ensemble des cardiopathies congénitales[1].
Il n'y a pas d'anomalie génétique spécifiquement liée à cette malformation et le plus souvent, le caryotype des sujets atteints est normal.
Certains médicaments ou toxiques sont incriminés à l'origine de cette anomalie : La prise de lithium pendant le premier trimestre de la grossesse augmenterait le risque, selon un facteur variable selon les auteurs mais pouvant atteindre 400 pour Weinstein & col[2].; un traitement maternel par benzodiazépine; une exposition maternelle aux vernis.
Embryologie
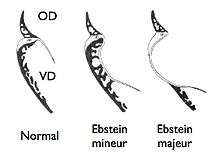
Normalement, l'appareil valvulaire tricuspide se forme par libération progressive de la couche interne du myocarde primitif, le clivage se faisant de la pointe du ventricule vers l'anneau valvulaire. La valve antérieure est totalement individualisée très précocement (embryon de 16 mm) alors que les feuillets postérieur et septal (ou médian) ne le sont que plus tardivement, après 3 mois.
La maladie d'Ebstein est la conséquence d'un défaut plus ou moins complet de ce clivage, qui n'atteint pas l'anneau. Le développement de perforations qui donnent lieu normalement à la formation des cordages et muscles papillaires est incomplet ou absent, d'où le caractère redondant de la valve.
Diagnostic
L'anomalie d'Ebstein est loin d'être univoque. Lorsque le défaut de clivage est minime, elle est souvent méconnue car sans aucun retentissement, le cœur fonctionnant dans des conditions quasi normales. À l'opposé, dans les formes majeures, la cavité ventriculaire droite est extrêmement réduite et incapable d'assurer la fonction qui lui est normalement dévolue. Entre ces deux extrêmes, tous les intermédiaires sont possibles.
Anténatal
Aspects échographiques avant la naissance
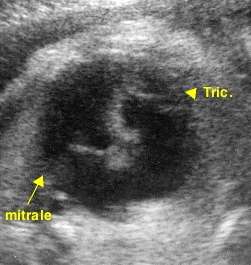
Le diagnostic peut être soupçonné sur l'incidence dite "des 4 cavités". Celle-ci permet d'analyser en particulier l'oreillette et le ventricule gauches (OG - VG), l'oreillette et le ventricule droits (OD - VD) et la position des valves mitrale (Mi) et tricuspide (Tr).
L'attention est attirée par une dilatation apparente de l'oreillette droite et l'impossibilité de visualiser, comme normalement, un appareil valvulaire tricuspide "symétrique" de l'appareil mitral. Cet appareil tricuspide est en fait retrouvé dans le ventricule droit, plus ou moins proche de son apex (pointe). Il est souvent difficile à distinguer de la "bandelette modératrice", structure musculaire physiologique traversant le ventricule droit.
L'examen en doppler-couleur permet d'apprécier l'existence et l'importance de la fuite valvulaire presque constamment associée.
- À titre pronostique, et outre l'importance du déplacement de la valve, l'examen recherche d'autres anomalies associées, en particulier :
- une voie d'éjection pulmonaire hypoplasique ou atrétique,
- une communication interventriculaire (CIV),
- des anomalies du cœur gauche,
- ...
Gestion de la grossesse
Contrairement à la plupart des malformations cardiaques, une malformation d'Ebstein dans une forme sévère peut être responsable non exceptionnellement d'une mort fœtale in utero. Dans la majorité des cas, la grossesse se poursuit néanmoins normalement et se posent alors deux questions :
- Quel en sera le pronostic à la naissance ? Peut-on envisager une interruption médicale de la grossesse ?
- quelle prise en charge à la naissance ?
Schématiquement, le pronostic néo-natal sera a priori bon dans les formes mineures et isolées. L'enfant peut même rester totalement asymptomatique et mener une vie tout à fait normale. Le seul risque serait celui de l'apparition de troubles du rythme cardiaque vis-à-vis desquels divers traitements habituellement sont efficaces. À l'opposé, on doit craindre le pire s'il s'agit d'une forme majeure ou s'il s'associe d'autres malformations, en particulier une communication inter-ventriculaire. Entre ces deux extrêmes, il est très difficile de poser un pronostic et les bonnes comme les mauvaises surprises ne sont pas rares. Cette incertitude fait qu'une interruption de grossesse peut être envisagée. Elle sera discutée au cas par cas.
Si la grossesse se poursuit, il est indispensable de prévoir l'accouchement (normal, par voie basse) à proximité d'un centre de réanimation néo-natal (c'est-à-dire dans une maternité "de niveau 3" en France). Le premier mois de vie représente en effet un cap particulièrement difficile à passer, nécessitant habituellement des mesures de réanimation hautement spécialisées.
Postnatal
Prise en charge du nouveau-né
Traitement
On ne peut parler de traitement curatif de l'anomalie d'Ebstein. Les diverses interventions possibles permettent de pallier la ou les complications. Les formes mineures de l'anomalie d'Ebstein ne demandant habituellement aucun traitement (hormis le traitement d'un éventuel trouble du rythme).
Traitement des troubles du rythme
C'est essentiellement celui des tachycardies supra-ventriculaires favorisées par la présence d'une ou plusieurs voies de pré-excitation (syndrome de Wolff-Parkinson-White).
- Médicaments anti-arythmiques ;
- ablation par cardiologie interventionnelle d'une ou plusieurs voie(s) de pré-excitation. Cette ablation est rendue difficile par l'anatomie particulière de l'oreillette d'une part, la fréquente multiplicité des voies accessoires d'autre part.
Traitement de la cyanose
Les formes associées à la présence d'une communication inter-auriculaire s'accompagnent d'une cyanose variable au repos mais constante et parfois intense au moindre effort, favorisée par le passage de sang désaturé de l'oreillette droite vers l'oreillette gauche (shunt droit-gauche). Cette cyanose (traduction d'une désaturation du sang artériel) limite fortement les capacités d'effort des sujets atteints et il est tentant de la supprimer en fermant cette communication entre les oreillettes. D'un autre côté, cette communication peut jouer un rôle bénéfique en proposant une "voie de décharge" au cœur droit et en retardant l'apparition de signes d'insuffisance cardiaque droite (œdèmes, gros foie ...).
La fermeture d'une ommunication inter-auriculaire n'est donc pas systématique mais ne sera envisagée qu'après qu'une "épreuve d'obstruction" (à l'aide d'une sonde muni d'un ballonnet obstruant de façon transitoire la communication) ait montré que cette obstruction ne s'accompagne pas d'une élévation excessive des pressions dans l'oreillette droite et les veines. Lorsqu'elle semble bénéfique, cette fermeture peut être pratiquée par voie chirurgicale ou, plus souvent actuellement, par mise en place d'une prothèse obstructive au cours d'un cathétérisme.
Traitement de l'insuffisance cardiaque droite
Outre le traitement médical commun à toute insuffisance cardiaque droite (reposant essentiellement sur les diurétiques), il peut être envisager deux types d'interventions :
- visant à améliorer le fonctionnement de la valve tricuspide malformée : Valvuloplastie chirurgicale (complexe et avec un résultat le plus souvent imparfait) plutôt que remplacement par une prothèse valvulaire qui s'accompagne d'un risque élevé de thrombose de valve, surtout chez l'enfant.
- visant à diminuer la quantité de sang transitant par l'oreillette et le ventricule droit : Dérivation cavo-pulmonaire partielle réalisée en créant une anastomose directe entre la veine cave supérieure et l'artère pulmonaire droite (type anastomose de Blalock-Taussig). Elle peut être accompagnée d'une exclusion du ventricule droit[3]. Ainsi, tout le sang revenant de l'extrémité supérieure du corps parvient directement aux poumons, sans passer par les cavités cardiaques qui en sont soulagées d'autant. Ce type d'intervention n'est envisageable qu'à la condition impérative que les résistances artérielles pulmonaires soient basses.
Conseil génétique
L'anomalie d'Ebstein n'est pas associée à une anomalie chromosomique spécifique. Quelques formes familiales ont été décrites. Il n'y a pas de conseil génétique spécifique à cette cardiopathie.
Historique
La première description date de 1866, faite par Wilhelm Ebstein (médecin allemand, Jawor 1836)[4] lors de l'autopsie d'un ouvrier âgé de 19 ans qui présentait une dyspnée, une cyanose et des palpitations depuis sa plus tendre enfance. Il faudra attendre 1949 pour que soit posé le premier diagnostic du vivant du malade. En 1958, Hernandez & coll décrivent une méthode diagnostique basée sur l'enregistrement simultané des pressions et de l'ECG endocavitaire. Ce procédé permet de mettre en évidence 3 "chambres" successives : le VD fonctionnel (Pression et ECG de type ventriculaire), le ventricule atrialisé (Pression auriculaire et ECG ventriculaire), l'OD proprement dite (pression et ECG de type auriculaire).
Références
- Rowe RD, Freedom RM, Mehrizi A. The neonate with congenital heart disease. 1981 Saunders, Philadelphia; p 515-528
- Weinstein MR. The International Register of Lithium Babies. Drug Information J 1976;10:94-100
- Reemtsen BL, Fagan BT, Wells WJ, Starnes VA, Current surgical therapy for Ebstein anomaly in neonates, J Thorac Cardiovasc Surg, 2006;132:1285–1290
- « The Two Anomalies of Wilhel Ebstein », Tex. Heart. Inst. J., vol. 44, no 3, , p. 198–201 (PMID 28761400, PMCID PMC5505398, DOI 10.14503/THIJ-16-6063)

