Techniques de comparaison des génomes
L’alignement de séquences est une pratique fondamentale pour de nombreuses applications de biologie comme la découverte de gènes et l’analyse phylogénétique.
Une nouvelle discipline est également née de la connaissance de ces séquences complètes de chromosomes, la génomique comparative. Il est maintenant possible de comparer deux organismes vivants à l’échelle de leur génome, de déterminer les gènes qu’ils ont en commun ou qui leur sont propres. Dans le contexte de l’identification sélective de gènes correspondant à des cibles thérapeutiques, en comparant par exemple une bactérie pathogène et une proche cousine non-pathogène, on peut essayer de repérer les gènes impliqués dans la virulence de la souche infectieuse. Ce nouveau domaine d’étude, traite les différents aspects de ce nouveau champ de la connaissance et s’appuie à la fois sur les concepts de la biologie que sur des outils issus de la chimie, de la physique et de l’informatique.
L’accélération du séquençage, permise en particulier par l'automatisation des méthodes d’analyse, nécessite un soutien de plus en plus important des technologies de l’informatique. Dans un premier stade, celui-ci est indispensable pour permettre l’assemblage de la « base de données » que constituent les milliers ou millions de fragments de génome. L’informatique est un outil incontournable pour extraire et analyser l’information contenue dans ces gigabases (1 Gbase =10^9 nucléotides) de séquence. Le volume des données à traiter est considérable. En 2002 les banques de séquences rassemblaient plus de 10^11 nucléotides et leur taille augmente exponentiellement. Les techniques d'accélération des comparaisons de génomes sont l’un des axes les plus importants en bio-informatique qui a pour but de remédier à un problème scientifique posé par la biologie « faire ressortir les régions ou séquences homologues ou différentes » ; on parle donc des méthodes de comparaison de deux ou plusieurs séquences de macromolécules biologiques (ADN, ARN ou protéines) les unes par rapport aux autres.
Analyse comparative des séquences génomiques
Dite aussi génomique comparative, elle consiste en l'étude comparative des structures et fonctions des différents génomes de différentes espèces en comparant une/des séquences génomiques aux séquences d'autres génomes, il s’agit donc de quantifier la similitude entre les séquences d'ADN/Protéines puis déterminer l'information contenue dans ces portions de génomes[1].
Les comparaisons peuvent se faire de multiples façons produisant différents types d'information[2] :
- par alignement (que ce soit l'alignement d'une portion d'un génome ou d'un génome complet) ;
- en comparant l'ordre de certains gènes ;
- en comparant la composition des séquences constituant le gènes ;
- etc.
Définitions
- Séquençage
Le séquençage de génome est l’un des champs d’application ou une sous-discipline de la bio-informatique, qui traite de l’analyse de données issues de l'information génétique contenue dans la séquence de l'ADN ou dans celle des protéines qu'il code. Cette branche s'intéresse en particulier à l'identification des ressemblances entre les séquences, à l'identification des gènes ou de régions biologiquement pertinentes dans l'ADN ou dans les protéines, en se basant sur l'enchaînement ou séquence de leurs composants élémentaires (nucléotides,acides aminés)[3].
- Séquence
Une Séquence génomique est l'enchaînement de molécules qui constituent une macromolécule, d'acide nucléique ou de protéine[4]. Elle est généralement représentée sous forme d'une chaîne de caractères stockée dans un fichier informatique au format texte utilisant (dans le cas d'une séquence d'ADN) l'alphabet des quatre lettres A, C, G et T, initiales des bases azotées - Adénine, Cytosine, Guanine et Thymine - qui distinguent les quatre types de nucléotides.
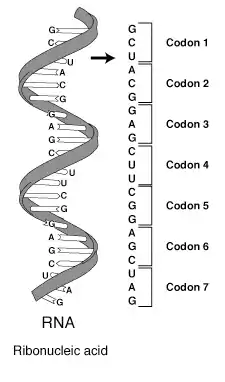
Et c'est l'enchaînement des vingt types d'acides aminés le long d'un polypeptide, classiquement représentée par une chaîne de caractères qui utilise un alphabet de vingt lettres dans le cas d'une séquence protéique[5].
Méthodes de séquençage
- méthode de Sanger (1975)[6],
- Méthode de séquençage Maxam–Gilbert (en)(1977)[7],
- Automatisation de Sanger (de ~1980 à 2005) au cours des dernières années, grâce à plusieurs avancées technologiques importantes dont l'utilisation de séquenceurs automatiques de gènes[8],[6],
- Nouvelles générations de séquenceurs (depuis 2005)
- Séquençage de l'ADN#Comparaison des méthodes de séquençage nouvelle-générationNGS : Séquençage de nouvelle génération[Note 1] (désormais largement utilisés) [9];
- HTS : Séquençage haut débit[Note 2],
- NNGS : Séquençage nouvelle-nouvelle Generation[Note 3] (en cours)

Analyse de séquences génomiques
Après la sélection d'un organisme, les projets génomiques partent sur trois procédures: le séquençage de l'ADN, l'assemblage de cette séquence pour créer une représentation du chromosome original, ainsi que l'annotation et l'analyse de celle-ci. L'analyse exploratoire peut être conduite soit sur la base de résultats expérimentaux soit par analogie avec des organismes modèles[11].
Les difficultés résident dans La disponibilité des données génomiques permettant de vérifier et/ou de tester beaucoup d'hypothèses et dans l’organisation de telle masses énormes d'informations pour offrir un accès aisé, à l'ensemble de la communauté des chercheurs, aux informations désirées. Cela a été rendu possible grâce à différentes bases de données, accessibles en lignes.

À l'échelle mondiale, trois grandes institutions sont chargées de l'archivage de ces données : le NCBI aux États-Unis « Centre national 'américain' pour les informations biotechnologiques », l'Institut européen de bio-informatique (EBI) en Europe et le DDBJ au Japon « Banque de données génétiques du Japon». Ces institutions se coordonnent pour gérer les grandes bases de données de séquences nucléotidiques comme GenBank ou l'EMBL database, ainsi que les bases de données de séquences protéiques comme UniProt ou TrEMBL.
Alors il est indispensable parfois, pour les chercheurs, d’exprimer leur besoin de logiciels et/ou d'algorithmes et de nouveaux outils d'analyse de séquences pour l’étude d’un problème donné, afin de pouvoir déterminer certaines propriétés, comme :
- La recherche d'une séquence dans une banque de données à partir d'une autre séquence ou d'un fragment de séquence. Les logiciels les plus fréquemment utilisés sont de la famille BLAST (blastn, blastp, blastx, tblastx et leurs dérivés),
- L'alignement de séquences pour trouver les ressemblances entre deux séquences et déterminer leurs éventuelles homologies. Les alignements sont à la base de la construction de parentés suivant des critères moléculaires, ou encore de la reconnaissance de motifs particuliers dans une protéine à partir de la séquence de celle-ci,
- La recherche de motifs ou structures qui permettent de caractériser les séquences,
- , etc.
Pour qu’ensuite, développer des Recherches sur les protéines à partir de la traduction de séquences nucléiques connues [12].
Comparaison de séquences
Dans la plupart des cas, le problème auquel l'utilisateur est confronté est formalisé comme suit: une nouvelle séquence est disponible et il est souhaitable de rechercher dans la base de données et de savoir si un ou des proches parents de cette séquence ont déjà été signalés. Si oui, on peut déduire par comparaison quelques-unes des données expérimentales recueillies de cette façon à la nouvelle séquence. Dans un tel cas, la solution consiste à comparer les séquences d'intérêt à toutes les séquences contenues dans la base de données, en gardant la trace de la plus semblable[13]. Deux outils très populaires Sont utilisés pour effectuer de telles recherches de similarité de base dans une base de données: FASTA et BLAST[14],[15],[16]. C'est par exemple l'une des idées développées dans la base de données PROSITE [16].
Alignement de séquences
Quand on parle de la comparaison de séquences on parle de l’Alignement qui est le processus par lequel deux ou plusieurs séquences sont comparées afin d'obtenir le plus de correspondances possibles entre les lettres qui les composent.
Les différents alignements sont :
- L'alignement local : consiste en l’alignement des séquences sur une partie de leur longueur,
- L'alignement global : consiste en l’alignement des séquences sur toute leur longueur,
- L'alignement optimal : consiste en l’alignement des séquences qui produit le plus haut score possible,
- L'alignement multiple : consiste en l’alignement global de trois séquences ou plus à la fois,
- Brèches ou gap : c’est un espace artificiel introduit dans une séquence pour contre-balancer et matérialiser une insertion dans une autre séquence. Il permet d'optimiser l'alignement entre les séquences[17],[18].
Annotation
L'annotation des génomes est une analyse informatique des séquences obtenues lors du séquençage permettant d'identifier les séquences informatives des génomes. Ces séquences sont principalement les gènes, on parle alors de prédiction de gènes. La plupart de ceux-ci sont identifiés soit par leur similitude avec des gènes déjà connus, soit par une prédiction en fonction de la séquence c'est-à-dire: présence d'un cadre de lecture ouvert caractérisée par un codon d'initiation de la traduction, puis au moins 100 codons et enfin un codon stop. Mais il existe aussi des « gènes morcelés » ou codons des ARN fonctionnels, ceux-ci doivent être prédits par des algorithmes différents.
Les gènes ne sont pas les seules cibles de l'annotation des génomes, il existe de nombreux autres types de séquences importantes dans les génomes, les séquences régulatrices, les éléments transposables, etc.[4],[19]
Objectif du séquençage et de la comparaison des séquences génomiques
Le séquençage du génome est une étape importante vers sa compréhension et la séquence du génome peut être considérée comme un raccourci du chemin aidant les scientifiques à trouver des gènes beaucoup plus facilement et rapidement. Une séquence génomique peut contenir même des indices sur l'endroit où se trouvent les gènes, comprendre comment le génome dans son ensemble fonctionne et comment les gènes travaillent ensemble pour diriger la croissance, le développement et le maintien d'un organisme entier[4],
En médecine, elle peut être utilisée pour identifier, diagnostiquer et potentiellement trouver des traitements à des maladies génétiques, en observant les mutations temporelles qui peuvent avoir des incidents sur les protéines et donc leurs rôles (rôles des protéines/fonction des protéines) et voir aussi dans quel gène elles apparaissent, permet d’induire les dysfonctionnements, identification des gènes spécifique à une espèce (Pathogénicité, ...), retrouver des régions de synténie (conservation de l'ordre de gènes homologues dans le génome d’espèces différentes)[20].
La comparaison de séquences est la tâche informatique la plus utilisée par les biologistes. Il s'agit de déterminer dans quelle mesure deux séquences, génomiques ou protéiques, se ressemblent.
La motivation première est d'inférer des connaissances sur une séquence à partir des connaissances attachées à une autre. Ainsi, si deux séquences sont très similaires et si l'une est connue pour être codante, l'hypothèse que la seconde le soit aussi peut être avancée. De même, si deux séquences protéiques sont similaires, il est souvent fait l'hypothèse que les protéines correspondantes assument des fonctions semblables ; si la fonction de l'une est connue, la fonction de la seconde peut ainsi s'en déduire.
Un biologiste qui détient une nouvelle séquence s'intéresse en premier temps à parcourir ces bases de données, à fin de trouver les séquences similaires et de faire hériter à la nouvelle séquence les connaissances qui leur sont associées. C'est également en comparant des séquences de génomes d'espèces actuelles qu'il est possible de reconstruire un arbre phylogénétique qui rend compte de l'histoire évolutive.
Il existe plusieurs bases de données qui contiennent l'ensemble des séquences nucléiques publiques avec leurs annotations (par exemple GenBank), ou l'ensemble des séquences protéiques expertisées (SwissProt)[5].
Profits tirés de la comparaison des génomes
- En médecine
- Aide à la création de nouveaux médicaments (prédiction de structure, d'interactions).
La greffe d'organes (ou transplantation d'organes) a pour but de remplacer un organe défaillant par un organe sain (cœur, foi , etc.) en cas de provenance externe, c'est-à-dire qu'il y a un donneur en question et on parle donc de l'allogreffe non pas de l'autogreffe,
- Recherche dans un laboratoire (entreprise publique, biotechs, pharmaceutique, , etc.).
- Aide à la création de tests et de systèmes de diagnostics destinés aux laboratoires d'analyses médicales, aux centres de transfusion sanguine et aux laboratoires de contrôle industriel, estimation de la probabilité et la rapidité de propagation des maladies.
- En science
- Étudier et déduire les différences entre les fonctionnements des cellules des différentes espèces,
- Étudier et comprendre l’être vivant[21].
- En informatique
- Développement de logiciels pour l'analyse et prédiction de données biologiques (génomique, transcriptomique, protéomique, etc.), par exemple la prédiction de gènes,
- Développement de logiciels pour la biologie : (LIMS, interface web, , etc..),
- Adaptation de technologies informatiques au domaine de la biologie,
- Nouvelle étude : reconstruction phylogénétique[22].
Algorithmes de comparaison des séquences génomiques
Méthodes de programmation dynamique
L'Algorithme Needleman-Wunsch est utilisé pour obtenir l'alignement global de deux séquences protéiques ou d'acides nucléiques et l'algorithme de Smith et Waterman est utilisé pour obtenir l'alignement local de deux séquences protéiques ou d'acides nucléiques[23].
En informatique, l'algorithme de Hirschberg (en), baptisé d'après son inventeur, Dan Hirschberg (en), est un algorithme de programmation dynamique qui trouve l'alignement optimal de séquences entre deux chaînes. L'optimalité est mesurée à l'aide de la distance de Levenshtein, définie comme étant la somme des coûts des insertions, des remplacements, des suppressions et des actions nulles nécessaires pour changer une chaîne par une autre. L'algorithme de Hirschberg est simplement décrit comme une version concurrente de l'algorithme Needleman-Wunsch[24]. Et il est couramment utilisé en bio-informatique pour trouver des alignements globaux maximaux de séquences d'ADN et de protéines.
Méthodes heuristiques
Ce sont des méthodes qui recherchent des similitudes dans une base de séquences[Note 5]. Les programmes des familles Fasta et BLAST sont des heuristiques qui réduisent le facteur temps en se basant sur l’idée de filtrage. Les deux simplifient le problème :
- en pré-sélectionnant les séquences de la banque susceptibles de présenter une similarité significative avec la séquence requête,
- et en localisant les régions potentiellement similaires dans les séquences.
Ces étapes sélectives permettent :
- de n'appliquer les méthodes de comparaison, coûteuses en temps, qu'à un sous-ensemble des séquences de la banque,
- et de restreindre le calcul de l'alignement optimal à des parties des séquences[18].
Le Programme FASTA[25],[15],[Note 6] ne considère que les séquences présentant une région de forte similitude avec la séquence recherchée. Il applique ensuite localement à chacune de ces meilleures zones de ressemblance un algorithme d'alignement optimal. La codification numérique des séquences, c'est-à-dire la décomposition de la séquence en courts motifs [Note 7] transcodés en entiers, confère à l'algorithme l'essentiel de sa rapidité.
Les programmes BLAST[15],[Note 8] « Recherche de Régions de Similarité Locales » sont une méthode heuristique qui utilise la méthode de Smith & Waterman. C'est un programme qui effectue un alignement local entre deux séquences nucléiques ou protéiques. La rapidité de BLAST permet la recherche des similarités entre une séquence requête et toutes les séquences d'une base de données.
Méthode d’apprentissage machine
L'apprentissage machine ou l'apprentissage automatique est un processus par lequel un ordinateur accroît ses connaissances et modifie son comportement à la suite de ses expériences et de ses actes passés. Cette méthode consiste en la conception, l'analyse, le développement et l'implémentation de méthodes permettant à une machine de réaliser des tâches difficiles que les algorithmiques classiques ne peuvent réaliser. Parmi les méthodes d'apprentissage machine :
- les réseaux de neurones ;
- les SVM [Note 9] machine à vecteur de support ;
- les k plus proches voisins ;
- l'algorithme espérance-maximisation EM[Note 10] ;
- le modèle de Markov caché[26].
Elles sont applicables dans plusieurs domaines tel que: la reconnaissance d'objets (visages, schémas, langages naturels, écriture, formes syntaxiques…) ; moteurs de recherche ; aide aux diagnostics, médical notamment, bio-informatique, chémoinformatique, , etc.
Accélération matérielle de la comparaison des séquences génomiques
Matériels de comparaison
Le traitement des données pour les applications de bio-informatique se fait actuellement par des logiciels, ce qui prend souvent beaucoup de temps, même aligner quelques centaines de séquences à l'aide d'outils d'alignement multiple consomme plusieurs heures CPU sur des postes de travail ultramodernes. L'analyse de séquences à grande échelle, qui implique souvent des dizaines de millions de séquences, est devenue un pilier, ainsi qu'un des principaux goulets d'étranglement dans la voie de la découverte scientifique. Le domaine de bio-informatique moléculaire héberge également un ensemble d'applications à forte intensité de calcul dans lesquelles les problèmes sous-jacents sont prouvés être intraitables en calcul (par exemple le calcul des arbres phylogénétiques, le repliement des protéines)[27].
En outre, des techniques de séquençage d'ADN à haut débit, qui ont permis de grandes avancées (séquençage complet du génome humain, projet d'annotation du génome des plantes) sont apparues. D’une autre vision ces progrès se sont traduits par le grand volume de données génomiques (ADN, protéines) disponibles pour la communauté, et qui est interprété par l'évolution des banques NCBI GenBank (pour l’ADN) UniProt (pour les protéines).
Les chercheurs se voient confrontés à un grand défi qui est l’extraction d’informations utiles à la compréhension de phénomènes biologiques, de ces volumes de données innombrables. Les outils classiques utilisés en bio-informatique ne sont pas conçus pour fonctionner sur de telles masses de données, et les volumes de calculs mis en jeu dans ces outils d'analyses sont devenus trop importants au point de devenir un goulot d'étranglement même pour les solutions offertes par l’informatique.
De nombreux travaux se sont donc intéressés à l'utilisation de machines parallèles pour réduire ces temps de calcul ; on parle alors de l'utilisation d'accélérateurs matériels spécialisés à base de logique programmable avec la possibilité de profiter des capacités d'accélération très élevées à consommation électrique réduite et des coûts de maintenance très raisonnables [28].
Pour accélérer les méthodes d'alignement des séquences, elles sont mises en œuvre sur diverses plates-formes matérielles disponibles[29], qui promettent un gain de performance énorme[30]. Et plusieurs accélérateurs matériels ont été proposés dont : SAMBA, FPGA, les GPU, les CPU, et ASIC[29].
GPU
Spécialisés pour des traitements synchrones de grosses quantités de données, les GPUs possèdent nativement une structure de cœurs massivement parallèle et offrent des puissances brutes de calcul largement supérieures aux processeurs[31]. Dans le domaine de la bio-informatique, les GPUs sont aussi prisés pour le traitement des séquences ADN. Avec quelques milliards de nucléotides, les GPUs permettent de réduire significativement les temps de traitements algorithmiques de ces chaînes, notamment pour les tris de très gros volumes de données (tris par base)[32].
FPGA
Les tendances récentes de la technologie informatique ont connu une progression rapide, comme les FPGA.
La mise en œuvre de la bio-informatique liée au FPGA et des applications de calculs en biologie est largement abordée [33].
L'augmentation de la densité et de la vitesse des circuits FPGA a ainsi favorisé l'émergence d'accélérateurs matériels reconfigurables orientés vers le domaine du calcul haute performance (HPC), avec plusieurs applications comme le calcul financier[34], grâce à sa fonctionnalité re-programmable, des développements de diverses applications biologiques sont possibles sur la même puce de silicium[29].
Ainsi ils se sont avérés être des architectures matérielles bien adaptées à la mise en œuvre de traitements de type bio-informatique[35].
La mise en œuvre FPGA utilise Xilinx Virtex II XC2V6000, une plate-forme pouvant accueillir 92 éléments de traitement avec une vitesse d'horloge maximale de 34 MHz[27]. Et sur les périphériques FPGA, la complexité d'une opération détermine directement la quantité consommée, de la surface de la puce [30].
ASIC
Un composant ASIC [Note 11] « circuit intégré propre à une application » est une puce dédiée à une seule fonction (ou à une classe restreinte de fonctions). Une fois conçu et fabriqué, il ne peut pas être modifié.
Dans les systèmes ASIC dédiés pour la comparaison de séquences, le calcul est généralement effectué par un réseau linéaire de processeurs ASIC identiques. La performance maximale de ces machines est impressionnante car tous les processeurs (quelques centaines) travaillent simultanément et de manière synchrone. La machine BioSCAN et la machine BISP appartiennent à cette catégorie. La puissance de calcul de ces machines dépend directement de la vitesse d'horloge et du nombre de processeurs[36].
En termes de vitesse, Il est reconnu qu'un ASIC est typiquement, 3 à 10 fois, plus rapide qu'un FPGA. Ainsi, on peut conclure que généralement les FPGAs peuvent fournir plus de vitesse que les processeurs, mais ne réalisent guère mieux les traitements que les ASIC. Le coût initial de conception et de production d'une unité FPGA est beaucoup plus faible que pour un ASIC, puisque le coût d'ingénierie non récurrente (NRE) d'un ASIC peut atteindre des millions de dollars. NRE représente le coût ponctuel correspondant à la conception et au test d'une nouvelle puce[37].
SAMBA
Le système SAMBA appartient à la catégorie ASIC, car le cœur du système est une matrice de processeurs VLSI dédiée, mais le système complet contient une interface de mémoire FPGA. Le réseau est connecté au poste de travail hôte par l'intermédiaire d'une carte mémoire FPGA qui agit comme un contrôleur de réseau et un mécanisme à grande vitesse pour alimenter correctement le réseau et filtrer les résultats à la volée.
La matrice du prototype SAMBA est composée de 32 puces identiques personnalisées, qui abritent chacune quatre processeurs, aboutissant à une matrice de processeurs. La puce a été conçue à IRISA et fournit une puissance de calcul de 400 millions d'opérations par seconde. Par conséquent, la matrice est capable d'atteindre 12,8 milliards d'opérations par seconde[36].
Processeurs
Les processeurs sont des architectures bien connues, souples et évolutives. En exploitant la répartition d'instructions SIMD extension de SSE montée sur les processeurs modernes, le temps de réalisation des analyses diminue de façon significative, ce qui rend les analyses de problèmes de données intensives, comme l'alignement des séquences, réalisables. De plus, les technologies émergentes du processeur comme le multi-cœur combinent deux processeurs indépendants ou plus.
Le paradigme du flux de données de multiples instructions simples(SIMD)[Note 12] est fortement utilisé dans cette classe de processeurs, ce qui le rend approprié pour les applications parallèles de données comme l'alignement des séquences. SIMD décrit des processeurs avec plusieurs éléments de traitement qui effectuent la même opération sur plusieurs données simultanément[38].
Historique
L'essor de cette discipline a été facilité par le développement des techniques de séquençage des génomes et la bio-informatique. En 1869, le Suisse Friedrich Miescher isole une substance riche en phosphore dans le noyau des cellules, qu'il nomme nucléine (le noyau). En 1896, l'Allemand Albrecht Kossel découvre dans l'acide nucléique les 4 bases azotées A, C, T, G. En 1928, Phoebus Levene et Walter Abraham Jacobs (en) (Etats-Unis) identifient le désoxyribose, et depuis 1935, on parle d'Acide désoxyribonucléique.
En 1944, l'américain Oswald Avery découvre que l'ADN est responsable de la transformation génétique des bactéries. Et certains scientifiques n'abandonnent pas l'idée que les protéines puissent porter l'information génétique.
Les expériences de Hershey et Chase confirment en 1952 l’hypothèse de l’ADN comme porteur de l'information génétique. En 1953 est publié dans Nature, par James Watson et Francis Crick une étude sur la structure de l'ADN en double hélice, grâce à la technique de diffraction des rayons X sur des cristaux de l'ADN, rendue possible par le travail de Rosalind Elsie Franklin.
Entre 1961 et 1965 le code génétique a été déchiffré « trois bases codent un acide aminé » (d'après la suggestion de George Gamow et l'expérience de Crick, Brenner et al., Philip Leder (en)). Il fallait travailler et chercher plusieurs années avant de pouvoir obtenir la première séquence de l’ADN,
En 1972, le premier véritable séquençage d'un génome est publié, avec la lecture de la séquence ARN du gène du virus Bacteriophage MS2 (en)[39]. Le projet de séquençage du génome humain (HGP)[Note 13] est un projet international lancé en 1990 aux États-Unis et coordonné par l'Institut national de senté (NIH)[Note 14] et par le département de l'Énergie [Note 15],
Craig Venter en 1998 annonce la création de l’entreprise Celera Genomics, en partenariat avec la multinationale PerkinElmer, spécialisée en électronique et leader mondial de l’équipement d’analyse de l’ADN. Cette même année, le HGP publie le GeneMap’98 qui contient 30 000 marqueurs.
En 1999, un premier chromosome humain est séquencé par une équipe coordonnée par le centre Sanger, en Grande-Bretagne. En , Celera Genomics annonce qu’elle détient dans sa banque de données 97 % des gènes humains, et propose les premiers résultats du séquençage total du génome humain. Et dans la même année, le HGP annonce 90 % du séquençage du génome humain. Les équipes scientifiques l'ont médiatisée par la compétition entre eux qui les a fait publier la première carte du génome humain, le à la fin du XXe siècle par Bill Clinton et Tony Blair.
En , les séquences du génome humain sont publiées par Nature (résultats du consortium public) et par Science (résultats de Celera Genomics).
Le , la fin du séquençage du génome humain est annoncée. Depuis, le séquençage évolue et le nombre de génomes complets séquencés.
En septembre 2007, une équipe menée par le biologiste et entrepreneur Craig Venter a publié le premier génome complet d'un individu qui est de Craig Venter lui-même.
Notes
- De l'anglais : Next Generation Sequencing.
- De l'anglais High-Throughput Sequencing.
- en: Next-Next Generation Sequencing.
- en: Single Molecule Sequencing.
- base de données génomiques ou banque de données.
- Basic Local Alignement Search Tool.
- nommés uplets.
- Basic Local Alignement Search Tool.
- support vector machine.
- Algorithme Expectation Maximisation.
- Application Spécificité Integrated Circuit.
- Single Instruction Multiple Data-Stream.
- HGP = Human Genome Project.
- NIH = le National Institute of Health.
- Department of Energy.
Références
- Thomas Derrien 2007, p. 2
- Catherine Matias 2015, p. 2
- Jean-Baptiste Waldner 2007, p. 121
- J. Craig Venter 2003
- François Rechenmann 2005
- F. Sanger 1977, p. 10
- Lilian T. C. França 2002, p. 183
- L.M. Smith 1986
- meth
- Equipe Bonsai 2014, p. 16
- Jonathan Pevsner 2009
- abi.snv
- Cédric Notredame 1998
- D. J. Lipman 1985
- S. F. Altschul 1990
- A. Bairoch 1997
- dsi.univ-paris5
- bioch
- EV. Koonin 2003
- Equipe Bonsai 2014, p. 74
- Equipe Bonsai 2014, p. 15
- batut 2014
- biochimej
- Kevin Wayne 2014, p. 9-20
- Pearson & Lipman 1988, p. 244
- Sonnhammer 1998, p. 320 - 322
- Souradip Sarkar 1988, p. 3790.
- Robert D. Stevens 2003, p. i302-i304.
- M.N. Isa 2011, p. 344.
- Thomas. B 2012, p. 169.
- Sidi Ahmed Mahmoudi, p. 1
- F. Sébastien 2010, p. 2
- B. Schmidt 2010
- G.L. Zhang 2006, p. 215-222
- Naeem Abbas 2013, p. 36
- P.Guerdoux 1997, p. 609-610
- Naeem Abbas 2012, p. 1-3
- Laiq Hasan, p. 189
- W. Min 1972, p. 82 - 88
Bibliographie
Analyse comparative des séquences génomiques
- Equipe Bonsai, « Cours d'introduction à la bioinformatique et de présentation des banques de séquences.1ère partie », Bioinformatique et données biologiques, , p. 16 (lire en ligne)
- Equipe Bonsai, « Cours d'introduction à la bioinformatique et de présentation des banques de séquences.1ère partie », Bioinformatique et données biologiques, , p. 74 (lire en ligne)
- Equipe Bonsai, « Cours d'introduction à la bioinformatique et de présentation des banques de séquences.1ère partie », Bioinformatique et données biologiques, , p. 15 (lire en ligne)
- Lilian T. C. Franca, Emanuel Carrilho et Tarso B. L. Kist, « A review of DNA sequencing techniques », Cambridge University Press, , p. 169–200 (DOI 10.1017/S0033583502003797, lire en ligne)
- Jean-Baptiste Waldner, « Nano-informatique et Intelligence Ambiante - Inventer l'Ordinateur du XXIe Siècle », Hermes Science, , p. 121 (ISBN 2-7462-1516-0)
- Thomas Derrien, « L'analyse comparée des génomes : applications à l'identification de nouveaux gènes canins », Bio-informatique [q-bio.QM]. Université Rennes 1, 2007., , p. 2 (ISBN 2-7462-1516-0, HAL tel-00656330)
- J. Craig Venter, « Genome Sequencing », Genome News Network is an editorially independent online,2000 - 2004 J. Craig Venter Institute., (lire en ligne)
- François Rechenmann, « Alignement optimal et comparaison de séquences génomiques et protéiques », l'équipe-projet IBIS,Explorez les sciences du numérique, (lire en ligne)
- F Sanger, G.M Air, B.G. Barrell, N.L. Brown, A.R. Coulson, C.A. Fiddes, C.A. Hutchison, P.M. Slocombe et M. Smith, « Nucleotide sequence of bacteriophage phi X174 DNA », Nature, vol. 265, , p. 687-695 (PMID 870828)
- Jonathan Pevsner, « Bioinformatics and functional genomics », Hoboken, N.J, Wiley-Blackwell, vol. 265, , p. 3-11 (ISBN 9780470085851, DOI 10.1002/9780470451496, lire en ligne)
- L.M. Smith, J.Z. Sanders, R.J. Kaiser, P. Hughes, C. Dodd, C.R. Conneell, C. Heiner, S.B. Kent et L.E. Hood, « Fluorenscence detection in automated DNA séquence analysis », Nature, vol. 321, , p. 674-679 (PMID 3713851)
- Cédric Notredame, « Use of genetic Algorithm for analysis of Biological Sequences », Université Paul Sabatier France, 2nd, , p. 15 (lire en ligne)
- Bairoch A, Bucher P et Hofmann K, « The Prosite database », Nucleic Acids Research, , p. 21-217
- Bérénice Batut, « Étude de l’évolution réductive des génomes bactériens par expériences d’évolution in silico et analyses bioinformatiques », Institut National des sciences appliquées de Lyon, , p. 23-213 (lire en ligne)
- Lipman D. J. et Pearson W. R., « Rapid and sensitive protein similarity searches », Science,1985.227, , p. 1435−1441
- Koonin EV et Galperin MY, « Sequence - Evolution - Function: Computational Approaches in Comparative Genomics-Chapter5:Genome Annotation and Analysis », NCBI-Boston: Kluwer Academic, (lire en ligne)
- Altschul S.F., Gish W, Miller W, Myers E.W. et Lipman D.J., « Basic local alignment search tool », J. Mol.Biol., , p. 215, 403 - 410
- (en) Purificación López-García et David Moreira, « Tracking microbial biodiversity through molecular and genomic ecology », Research in Microbiology, vol. 159, no 1, , p. 67–73 (DOI 10.1016/j.resmic.2007.11.019)
- Catherine Matias, « II. Génomique comparative », CNRS - Laboratoire de Probabilités et Modèles Aléatoires, Paris, , p. 1 (lire en ligne)
- « L’analyse des génomes complets » (consulté en )
- « Algorithmes et programmes de comparaison de séquences Interprétation des résultats : E-value, P-value », 2001-2017 (consulté en )
- « Génomique : les méthodes de séquençage d'acides nucléiques et l'acquisition des données » (consulté en )
Algorithmes de comparaison des séquences génomiques
- « Génomique : les méthodes de séquençage d'acides nucléiques et l'acquisition des données », sur biochimej.univ-angers.fr, 2001-2017
- Kevin Wayne, « 6.Dynamic ProgrammingII,6.6-Hirschberg's Algorithm », 2005 Pearson-Addison Wesley, , p. 1-50 (lire en ligne)
- Pearson W.R et Lipman D.J, « Improved tools for biological sequence comparison », Proc. Natl. Acad. Sci. USA. 85, , p. 244
- « fam: multiple sequence alignments and HMM-profiles of protein domains », Nucleic Acids Res. 26, , p. 320 - 322
Accélération matériel de la comparaison des séquences génomiques
- « multiple sequence alignments and HMM-profiles of protein domains" Nucleic Acids », Pfam, , p. 320 - 322
- « Improved tools for biological sequence comparison », Proc. Natl. Acad. Sci.,
- Souradip Sarkar,Turbo Majumder, Ananth Kalyanaraman, Partha Pratim Pande, « Hardware Accelerators for Biocomputing: A Survey », School Of Electrical Engineering and Computer Science,Washington State University, Pullman, USA, , p. 3789-5036 (lire en ligne)
- Thomas B,Preußer and Oliver Knodel and Rainer G. Spallek, « Short-Read Mapping by a Systolic Custom FPGA Computation », IEEE ComputerSociety, , p. 169 (DOI 10.1109/FCCM.2012.37)
- Isa M.N,K. Benkrid, T. Clayton, C. Ling, and A.T. Erdogan, « An FPGA-based Parameterised and Scalable, Optimal Solutions for Pairwise Biological Sequence Analysis », School of Engineering, The University of Edinburgh, Edinburgh,NASA/ESA Conference on Adaptive Hardware and Systems, , p. 344
- Sidi Ahmed Mahmoudi,Sébastien Frémal, Michel Bagein, Pierre Manneback, « Calcul intensif sur GPU:exemples en traitement d’images, en bioinformatique et en télécommunication », Université de Mons, Faculté Polytechnique Service d’informatique, ***, p. 1
- Sébastien F., « Conception et mise en œuvre d’algorithmes de sélection de ressources dans un environnement informatique hétérogène multiprocesseur », Rapport de Travail de Fin d'Etude, , p. 2
- Schmidt B, H.Schroder, and M. Schimmler, « Massively parallel solutions for molecular sequence analysis », IPDPS,
- Isa M.N,K. Benkrid, T. Clayton, C. Ling, and A.T. Erdogan, « An FPGA-based Parameterised and Scalable, Optimal Solutions for Pairwise Biological Sequence Analysis », School of Engineering, The University of Edinburgh, Edinburgh,NASA/ESA Conference on Adaptive Hardware and Systems, , p. 344
- Souradip Sarkar,Turbo Majumder, Ananth Kalyanaraman, Partha Pratim Pande, « Hardware Accelerators for Biocomputing: A Survey », School Of Electrical Engineering and Computer Science,Washington State University, Pullman, USA, , p. 3790 (lire en ligne)
- Guerdoux-Jamet P,D.Lavenier, « SAMBA: hardware accelerator for biological sequence comparison », IRISA, Campus de Beaulieu, , p. 609-610 (DOI 10.1109/FCCM, lire en ligne)
- Naeem Abbas, « Acceleration of a bioinformatics application using high-level synthesis », École normale supérieure de Cachan - ENS Cachan, , p. 36 (lire en ligne)
- Laiq HasanAl-Ars, « An Overview of Hardware-Based Acceleration of Biological Sequence Alignment », TU Delft The Netherlands, ***, p. 189 (lire en ligne)
- JRobert D.Stevens, Alan J.Robinson et Carole A.Goble, « myGrid: personalised bioinformatics on the information grid », Revue Bioinformatics, vol. 19, , i302-i304 (bioinformatics.oxfordjournals.org/content/19/suppl_1/i302.full.pdf)
- Naeem Abbas, « Acceleration of a bioinformatics application using high-level synthesis », École normale supérieure de Cachan - ENS Cachan, , p. 1-3 (HAL tel-00847076/document)
- G.L. Zhang, P.H.W. Leong, C.H. Ho, K.H. Tsoi, C.C.C. Cheung, D.U. lee, R.C.C. Cheung et W. Luk, « Reconfigurable acceleration for Monte Carlo based financial simulation », Field-Programmable Technology, 2005. Proceedings. 2005 IEEE International Conference, , p. 215-222 (ISBN 0-7803-9407-0, DOI 10.1109/FPT.2005.1568549id=1111, lire en ligne)
Historique
- W. Min Jou, G Haegeman, M Ysebaert et W Fiers, « Nucleotide Sequence of the Gene Coding for the Bacteriophage MS2 Coat Protein », Nature 237,Laboratory of Molecular Biology and Laboratory of Physiological Chemistry, State University of Ghent, Belgium, , p. 82 - 88 (DOI 10.1038/237082a0, lire en ligne)
Autres Sources
- « Sequence - Evolution - Function: Computational Approaches in Comparative Genomics. » (consulté le )
- « alphabet de vingt lettres »
- Haruo Ikeda1, Jun Ishikawa, Akiharu Hanamoto, Mayumi Shinose, Hisashi Kikuchi, Tadayoshi Shiba,Yoshiyuki Sakaki, Masahira Hattori1,and Satoshi O ¯mura, « Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis », Researcharticle, (DOI 10.1038/nbt820, lire en ligne)
- Valentin Wucher, « Modélisation d’un réseau de régulation d’ARN pour prédire des fonctions de gènes impliques dans le mode de reproduction du puceron du pois », Submitted on 26 Mar 2015, (HAL tel-01135870)
- « Classification et caractérisation de familles enzymatiques a l’aide de méthodes formelles Gaelle Garet », Submitted on 2 Feb 2015, (HAL tel-01096916v2)
- « Recherche de similarités dans les sequences d’ADN : modeles et algorithmes pour la conception de graines efficaces », Département de formation doctorale en informatique École doctorale IAEM Lorraine UFR STMIA,
- Thomas Derrien, « L’analyse comparée des génomes : applications `a l’identification de nouveaux gènes canins. », Bio-informatique [q-bio.QM]. Université Rennes 1, (HAL tel-00656330)
Voir aussi
- Séquence (acide nucléique)
- Séquençage de l'ADN
- Séquençage de l'ARN ou RNA-Seq
- Séquenceur d'ADN
- Séquencement complet du génome (en)
- Bio-informaticien
- Biologie
- Biologie de synthèse
- Biotechnologie
- Informatique
- Nanotechnologie
- Nanotechnologies
- biotechnologies
- informatique et sciences cognitives
- NBIC
- Séquence (acide nucléique)
Organismes
Liens externes
- Questions à propos du séquençage du génome humain
- La bio-informatique : Annexe 2 (version archivée) (Le centre de ressources Infobiogen a cessé ses activités en )
- Société française de bioinformatique (SFBI ; société savante créée en 2005 par des chercheurs et enseignants-chercheurs en bio-informatique).
- Institut suisse de bioinformatique (ISB ; institut créé en 1998 pour regrouper les chercheurs en bio-informatique en Suisse).
- Bioinfo-fr (Bioinfo-fr.net ; Blog communautaire scientifique conçu par des bio-informaticien(ne)s francophones).
- JeBiF (JeBiF ; association des jeunes bio-informaticiens de France créée en 2008).
- Sélection de sites web sur la bio-informatique dans le répertoire encyclopédique : Les Signets de la Bibliothèque nationale de France
 Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire  Portail de l'informatique théorique
Portail de l'informatique théorique  Portail de la linguistique
Portail de la linguistique