Syndrome de Silver-Russell
Décrit pour la première fois en 1953[2],[3], le syndrome de Silver-Russell (SSR) associe un retard de croissance commençant en période fœtale et se continuant en période postnatale. La taille et le poids à la naissance se situent en dessous du cinquième centile. Les enfants atteints ont un nanisme harmonieux, un périmètre crânien normal, une clinodactylie du cinquième doigt, un dymorphisme facial avec très souvent une asymétrie de croissance d'un hémi-corps. Ces enfants ont une augmentation du risque de retard de développement avec des capacités d'apprentissages réduites.
| Syndrome de Silver-Russell | |
| Référence MIM | 180860 |
|---|---|
| Transmission | Voir l'article |
| Chromosome | 11 11p15 |
| Empreinte parentale | oui |
| Mutation | Voir l'article |
| Porteur sain | Sans objet |
| Incidence | 1 sur 100 000 naissances[1] |
| Nombre de cas | 500 cas connus |
| Maladie génétiquement liée | Aucune |
| Diagnostic prénatal | Possible |
| Liste des maladies génétiques à gène identifié | |
Ce syndrome est plus un phénotype qu'une maladie génétiquement homogène. Le diagnostic repose essentiellement sur les signes cliniques: une petite taille avec un périmètre crânien normal. L'enquête génétique sera normale dans 40 % des individus atteint par ce syndrome
Causes
Les causes génétiques du syndrome de Russell-Silver sont complexes. Le trouble résulte souvent de la régulation anormale de certains gènes qui contrôlent la croissance. La recherche s'est concentrée sur les gènes situés dans des régions particulières du chromosome 7 et du chromosome 11. Les gens héritent normalement d'une copie de chaque chromosome de leur mère et d'une copie de leur père. Pour la plupart des gènes, les deux copies sont exprimées ou «activées» dans les cellules. Pour certains gènes, cependant, seule la copie héritée du père d'une personne (la copie paternelle) est exprimée. Pour les autres gènes, seule la copie héritée de la mère d'une personne (la copie maternelle) est exprimée[4].
Ces différences d'expression génique propres aux parents sont causées par un phénomène appelé empreinte génomique. Le chromosome 7 et le chromosome 11 contiennent tous deux des groupes de gènes qui subissent normalement une empreinte génomique; certains de ces gènes ne sont actifs que sur la copie maternelle du chromosome, tandis que d'autres ne sont actifs que sur la copie paternelle. Les anomalies impliquant ces gènes semblent être responsables de nombreux cas de syndrome de Russell-Silver[4]..
Les chercheurs soupçonnent que 30 à 50 % de tous les cas de syndrome de Russell-Silver résultent de changements dans un processus appelé méthylation sur le bras court (p) du chromosome 11 en position 15 (11p15). La méthylation est une réaction chimique qui attache de petites molécules appelées groupes méthyle à certains segments de l'ADN. Dans les gènes qui subissent une empreinte génomique, la méthylation est une façon dont le parent d'origine d'un gène est marqué pendant la formation des ovules et des spermatozoïdes. Le syndrome de Russell-Silver a été associé à des changements de méthylation impliquant les gènes H19 et IGF2, qui sont situés les uns à côté des autres à 11p15. On pense que ces gènes sont impliqués dans la direction d'une croissance normale. Une perte de méthylation perturbe la régulation de ces gènes, ce qui entraîne une croissance lente et les autres caractéristiques de ce trouble[4]..
Des anomalies impliquant des gènes sur le chromosome 7 peuvent également provoquer le syndrome de Russell-Silver. Dans 7 à 10 % des cas, les gens héritent des deux copies du chromosome 7 de leur mère au lieu d'une copie de chaque parent. Ce phénomène est appelé disomie uniparentale maternelle (UPD). L'UPD maternelle fait que les gens ont deux copies actives de certains gènes imprimés et aucune copie active d'autres. Un déséquilibre de certains gènes paternels et maternels actifs sur le chromosome 7 sous-tend les signes et symptômes de la maladie.Chez environ 40% des personnes atteintes du syndrome de Russell-Silver, la cause de la maladie est inconnue. Il est probable que des changements impliquant des gènes imprimés sur des chromosomes autres que 7 et 11 jouent un rôle[4].
L'hypométhylation de la région de contrôle 1 et 2 (gène H19) au niveau de situé 11p15,5 provoque une SSR chez 35 à 50 % des individus, et une disomie uniparentale maternelle chromosome 7 humain[5]. provoque une SRS chez 7 à 10 % des individus.
Il y a un petit nombre de personnes atteintes de SSR qui ont des duplications, des suppressions ou des translocations impliquant les centres d'impression à 11p15,5 ou des duplications, des suppressions ou des translocations impliquant le chromosome 7.
Description
Le syndrome de Russell-Silver est un trouble de la croissance caractérisé par une croissance lente avant et après la naissance. Les bébés atteints de cette affection ont un faible poids à la naissance et ne parviennent souvent pas à grandir et à prendre du poids au rythme attendu (retard de croissance). La croissance de la tête est cependant normale, de sorte que la tête peut sembler inhabituellement grande par rapport au reste du corps. Les enfants atteints sont minces et ont un faible appétit, et certains développent des épisodes récurrents d'hypoglycémie à la suite de difficultés d'alimentation. Les adultes atteints du syndrome de Russell-Silver sont petits : la taille moyenne pour les hommes affectés est d'environ 151 centimètres et la taille moyenne pour les femmes touchées est d'environ 140 centimètres[4].
De nombreux enfants atteints du syndrome de Russell-Silver ont un petit visage triangulaire avec des caractéristiques faciales distinctives, notamment un front proéminent, un menton étroit, une petite mâchoire et des coins de la bouche baissés. D'autres caractéristiques de ce trouble peuvent inclure une courbure inhabituelle du cinquième doigt (clinodactylie), une croissance asymétrique ou inégale de certaines parties du corps et des anomalies du système digestif. Le syndrome de Russell-Silver est également associé à un risque accru de retard de développement, de problèmes de parole et de langage et de troubles d'apprentissage[4].
Le prévalence de ce syndrome pourrait atteindre 1 sur 16 000 naissances[6]
Critères morphologiques
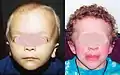 Contour du visage, vu de face, de forme triangulaire, avec des tempes larges se rétrécissant en un menton étroit.
Contour du visage, vu de face, de forme triangulaire, avec des tempes larges se rétrécissant en un menton étroit. Une phalange qui est incurvée latéralement dans le plan de la paume.
Une phalange qui est incurvée latéralement dans le plan de la paume. Commissures buccales positionnées en dessous de la fissure labiale médiane.
Commissures buccales positionnées en dessous de la fissure labiale médiane. Longueur et largeur de la mandibule apparemment réduites vu de face mais pas de côté.
Longueur et largeur de la mandibule apparemment réduites vu de face mais pas de côté. Distance bigoniale (largeur faciale inférieure) plus de 2 dérivations standards en dessous de la médiane.
Distance bigoniale (largeur faciale inférieure) plus de 2 dérivations standards en dessous de la médiane. Proéminence vers l'avant de tout le front, en raison de la saillie de l'os frontal.
Proéminence vers l'avant de tout le front, en raison de la saillie de l'os frontal.
Diagnostic
Le syndrome de Silver-Russell est une maladie génétiquement hétérogène ; le diagnostic clinique nécessite le respect de critères cliniques spécifiques décrits dans le système de notation clinique Netchine-Harbison (NH-CSS)[7]. Ce système de notation a été accepté comme méthode pour identifier les personnes qui devraient subir des examens complémentaires pour le diagnostic de SSR[8]
Clinique
Si un individu répond à quatre des six critères, le diagnostic clinique est suspecté et un test de confirmation moléculaire est justifié. Quelques rares individus répondant à trois des six critères ont eu une confirmation moléculaire positive pour le SSR.
| Critères 2017 de suspicion de SSR |
|---|
| Petit pour l'âge gestationnel (poids ou taille à la naissance en dessous de 2 dérivations standards par rapport à la médiane pour l'âge gestationnel) |
| Retard de croissance postnatal (poids ou taille en dessous du 25e percentile à 2 ans |
| Macrocéphalie relative à la naissance (circonférence de la tête supérieure à 1,5 dérivation standard du périmètre crânien pour l'âge gestationnel |
| Front proéminent (front dépassant du plan facial sur une vue latérale entre 1 et 3 ans) |
| Asymétrie corporelle (écart de longueur de membre supérieure à 0,5 cm ou inférieure à 0,5 cm avec plus de 2 autres parties du corps asymétriques) |
| Difficultés d'alimentation ou indice de masse corporelle inférieur à 2 dérivations standards à 24 mois ou utilisation actuelle d'une sonde d'alimentation ou usage de la cyproheptadine pour stimuler l'appétit |
Diagnostic moléculaire
Il faut rechercher une disomie uniparentale maternelle au niveau du chromosome 7 ou une régulation anormale de la transcription de deux régions soumis à empreinte sur le chromosome 11p15.5 : ICR1 et ICR2. La régulation peut être perturbée par de nombreux mécanismes.Il existe d'autres méthodes de diagnostic très élaboré si aucune de ses deux méthodes est concluantes.
Environ 40 % des individus qui subissent des tests moléculaires ayant au moins 4 critères cliniques auront des résultats moléculaires négatives après avoir employé toutes les méthodes possibles. Pour les individus de ce groupe, un diagnostic clinique de SSR peut être établi si : deux des quatre signes cliniques identifiés sont des bosses frontales / frontales proéminentes et une macrocéphalie relative à la naissance ; et d'autres maladies génétiques ont été exclues[9].
Gènes pouvant être impliqués dans ce syndrome
Si l'analyse de méthylation des études 11p15.5 ICR1 / ICR2 et UPD7 est normale, un panel multigène qui comprend l'analyse de séquence d'IGF2, CDKN1C, PLAG1, HMGA2 et d'autres gènes peut révéler des anomaliess[9].
Troubles génétiquement apparentés (alléliques)
| Altération moléculaire | Phénotype |
|---|---|
| Régulation anormale de la transcription des gènes soumis à empreinte parental sur le 11p15.5 ou par héritage maternelle de la perte de fonction dans CDKN1C | Syndrome de Beckwith-Wiedemann |
| Altérations moléculaires du 11p15, y compris hypométhylation au niveau de ICR2 ou hyperméthylation au niveau de ICR1 ou 2 et 11p15 parental disomie paternelle | Hémi-hypertrophie corporéale |
| Mosaïcisme somatique par hypométhylation de l'ICR1 paternel | Hémi-hypertrophie corporéale |
| Altérations constitutionnelles de 11p15.5 y compris l'hyperméthylation de l'ICR1, disomie parentale paternel de 11p15.5, et anomalies génomiques y compris microdélétion et microinsertion | Tumeur de Wilms |
| Variante pathogène héritée de la mère dans CDKN1C | Syndrome IMAGe |
Diagnostic différentiel
Il se pose devant tout retard de croissance intra-utérin ou de nanisme.
| Pathologie | Gènes | Mode de transmission |
|---|---|---|
| Maladies génétiques | ||
| Syndrome 3 M | CCDC8
CUL7 OBSL1 |
Autosomique récessif |
| Syndrome de Bloom | BLM | Autosomique récessif |
| Maladie de Fanconi | Plus de 20 gènes | Autosomique récessif
Autosomique dominant Lié au chromosome X |
| Syndrome de Nimègue | NBN | Autosomique récessif |
| Syndrome IMAGe | CDKN1C | Autosomique récessif |
| Syndrome de cassure chromosomique de Varsovie | DDX11 | Autosomique récessif |
| Syndrome de Meier-Gorlin | CDC45
CDC6 CDT1 GMNN MCM5 ORC1 ORC4 ORC6 |
Autosomique récessif
Autosomique dominant |
| Résistance au facteur de croissance 2 ressemblant à l'insuline | IGF1R | Autosomique récessif
Autosomique dominant |
| Maladies chromosomiques | ||
| Diploïdie/Triploïdie | ||
| Syndrome de Temple | Autosomique dominant | |
| Intoxications fœtales | ||
| Exposition prénatale à l'alcool | ||
Mode de transmission
La plupart des cas de syndrome de Russell-Silver sont sporadiques, ce qui signifie qu'ils surviennent chez des individus sans antécédents de trouble dans leur famille.
Rarement, le syndrome de Russell-Silver affecte une famille. Dans certaines familles touchées, la maladie semble avoir un mode de transmission autosomique dominant. L'hérédité autosomique dominante signifie qu'une copie d'un changement génétique dans chaque cellule est suffisante pour provoquer le trouble. Dans d'autres familles, l'affection semble avoir un mode de transmission autosomique récessif. L'hérédité récessive autosomique signifie que les deux copies d'un gène sont modifiées dans chaque cellule. Les parents d'un individu ayant une maladie autosomique récessive portent chacun une copie du gène muté, mais ils ne présentent généralement pas de signes et de symptômes de la maladie.
Les syndromes de Russell-Silver par disomie uniparentale sont des cas uniques dans la famille. Les autres syndromes de Russell-Silver ont des modes de transmission évoquant soit un mode autosomique ou gonosomique à la fois récessif ou dominant.
Grossesse chez une femme atteinte de ce syndrome
Une grossesse spontanée avec diabète gestationnel et un accouchement sans problème ont été décrits chez une femme chinoise atteinte du syndrome de Silver-Russell[10].
Conseil génétique
Le conseil génétique est très difficile en raison de l'hétérogénéité de syndrome.
Diagnostic prénatal
Si le SSR est due à une disomie uniparentale maternelle celle-ci peut être diagnostiquée avant la naissance par ponction de sang fœtal[9].
Si une anomalie chromosomique causale (par exemple, une translocation déséquilibrée impliquant 11p15.5, une microdélétion paternelle ou une microduplication maternelle du chromosome 11p15.5 incluant CDKN1C) a été identifiée chez la mère, un diagnostic prénatal sur des cellules fœtales obtenu par ponction de sang fœtal ou l'amniocentèse est possible[9].
Liens externes
- Site en français de renseignement sur les maladies rares et les médicaments orphelins
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number:18086
- (en)Saal HM, Harbison MD, Netchine I. « Silver-Russell Syndrome ». 2002 Nov 2 [Updated 2019 Oct 21]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020[11].
Notes et références
- (en) Christoforidis A, Maniadaki I, Stanhope R (2005) « Managing children with Russell-Silver syndrome: more than just growth hormone treatment? » J Pediatr Endocrinol Metab. 18:651-2
- (en) Silver HK, Kiyasu W, George J, Deamer WC (1953) « Syndrome of congenital hemihypertrophy, shortness of stature, and elevated urinary gonadotropins » Pediatrics 12:368-76.
- (en) Russell A (1954) « A syndrome of intra-uterine dwarfism recognizable at birth with cranio-facial dysostosis, disproportionately short arms, and other anomalies (5 examples) » Proc R Soc Med. 47:1040-4
- (en) National Library of Medicine (US). Genetics Home Reference [Internet]. Bethesda (MD), « Russell-Silver syndrome », sur ghr.nlm.nih.gov, (consulté le )
- (en) Eggermann T, Spengler S, Gogiel M, Begemann M, Elbracht M, « Epigenetic and genetic diagnosis of Silver-Russell syndrome » Expert Rev Mol Diagn, 2012;12:459-471
- (en) Maria Yakoreva, Tiina Kahre, Riina Žordania et Karit Reinson, « A retrospective analysis of the prevalence of imprinting disorders in Estonia from 1998 to 2016 », European Journal of Human Genetics, vol. 27, no 11, , p. 1649–1658 (ISSN 1018-4813 et 1476-5438, PMID 31186545, PMCID PMC6871525, DOI 10.1038/s41431-019-0446-x, lire en ligne, consulté le )
- (en) Salah Azzi, Jennifer Salem, Nathalie Thibaud et Sandra Chantot-Bastaraud, « A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver-Russell syndrome », Journal of Medical Genetics, vol. 52, no 7, , p. 446–453 (ISSN 0022-2593 et 1468-6244, PMID 25951829, PMCID PMC4501172, DOI 10.1136/jmedgenet-2014-102979, lire en ligne, consulté le )
- (en) Wakeling, E., Brioude, F., Lokulo-Sodipe, O. et al. Diagnosis and management of Silver–Russell syndrome: first international consensus statement. Nat Rev Endocrinol 13, 105–124 (2017). DOI:10.1038/nrendo.2016.138
- (en) Saal HM, Harbison MD, Netchine I. « Silver-Russell Syndrome ». 2002 Nov 2 [Updated 2019 Oct 21]. In: Adam MP, Ardinger HH, Pagon RA et al. editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1324/
- (en) Mengte Shi, Luya Ruan, Xiaomin Shi et Yaoxin Zhu, « A spontaneous pregnancy and successful delivery in a Chinese female with Silver-Russell syndrome accompanied by gestational diabetes mellitus », Gynecological Endocrinology, vol. 35, no 9, , p. 752–755 (ISSN 0951-3590 et 1473-0766, DOI 10.1080/09513590.2019.1590547, lire en ligne, consulté le )
- Howard M. Saal, Madeleine D. Harbison et Irene Netchine, « Silver-Russell Syndrome », dans GeneReviews®, University of Washington, Seattle, (PMID 20301499, lire en ligne)
 Portail de la médecine
Portail de la médecine  Portail du handicap
Portail du handicap