Séquençage de l'ADN
Le séquençage de l'ADN consiste à déterminer l'ordre d'enchaînement des nucléotides pour un fragment d’ADN donné.

La séquence d’ADN contient l’information nécessaire aux êtres vivants pour survivre et se reproduire. Déterminer cette séquence est donc utile aussi bien pour les recherches visant à savoir comment vivent les organismes que pour des sujets appliqués. En médecine, elle peut être utilisée pour identifier, diagnostiquer et potentiellement trouver des traitements à des maladies génétiques et à la virologie. En biologie, l'étude des séquences d'ADN est devenue un outil important pour la classification des espèces.
Historique


Le séquençage de l'ADN est inventé dans la deuxième moitié des années 1970. Deux méthodes sont développées indépendamment, l'une par l'équipe de Walter Gilbert, aux États-Unis, et l'autre par celle de Frederick Sanger (en 1977), au Royaume-Uni. Ces deux méthodes sont fondées sur des principes diamétralement opposés : l'approche de Sanger est une méthode par synthèse enzymatique sélective, tandis que celle de Maxam et Gilbert est une méthode par dégradation chimique sélective. Pour cette découverte, Gilbert et Sanger sont récompensés par le prix Nobel de chimie en 1980.
Initialement, la méthode de Sanger nécessitait de disposer d'un ADN simple brin qui servait de matrice pour la synthèse enzymatique du brin complémentaire. Pour cette raison, le premier organisme biologique dont le génome a été séquencé en 1977 est le virus bactériophage φX174[1]. Ce virus a la propriété d'avoir un génome constitué d'ADN simple brin qui est encapsulé dans la particule virale.
Au cours des 25 dernières années, la méthode de Sanger a été largement développée grâce à plusieurs avancées technologiques importantes :
- la mise au point de vecteurs de séquençage adaptés, comme le phage M13 développé par Joachim Messing au début des années 1980[2] ;
- le développement de la synthèse chimique automatisée des oligonucléotides qui sont utilisés comme amorces dans la synthèse ;
- l'introduction de traceurs fluorescents à la place des marqueurs radioactifs utilisés initialement. Ce progrès a permis de sortir le séquençage des pièces confinées nécessaires à l'usage de radio-isotopes[3] ;
- l'adaptation de la technique PCR pour le séquençage ;
- l'utilisation de séquenceurs automatiques de gènes[3] ;
- l'utilisation de l'électrophorèse capillaire pour la séparation et l'analyse[4].
La méthode de Maxam et Gilbert nécessite des réactifs chimiques toxiques et reste limitée quant à la taille des fragments d'ADN qu'elle permet d'analyser (< 250 nucléotides). Moins facile à robotiser, son usage est devenu aujourd'hui confidentiel.
Méthodes
Méthode de Sanger (1977)

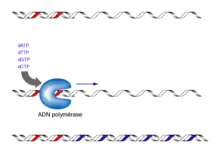
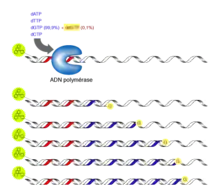
Cette méthode est utilisée classiquement pour effectuer un petit séquençage ponctuel. Pour le séquençage d’un génome entier, on utilise plutôt le séquençage nouvelle génération. Le principe de cette méthode consiste à initier la polymérisation de l’ADN à l'aide d'un petit oligonucléotide (amorce) complémentaire à une partie du fragment d’ADN à séquencer. L’élongation de l’amorce est réalisée par le fragment de Klenow (une ADN polymérase I dépourvue d’activité exonucléase 5’→3’) et maintenue par des ADN polymérases thermostables, celles qui sont utilisées pour la PCR. Les quatre désoxyribonucléotides (dATP, dCTP, dGTP, dTTP) sont ajoutés, ainsi qu’une faible concentration de l'un des quatre didésoxyribonucléotides (ddATP, ddCTP, ddGTP ou ddTTP)[5].
Ces didésoxyribonucléotides agissent comme des « poisons » terminateurs de chaîne : une fois incorporés dans le nouveau brin synthétisé, ils empêchent la poursuite de l’élongation car ils ne possèdent pas d'extrémité 3'-OH (seulement un hydrogène à la place du groupement hydroxyle). Cette terminaison se fait spécifiquement au niveau des nucléotides correspondant au didésoxyribonucléotide incorporé dans la réaction. Pour le séquençage complet d'un même fragment d'ADN, on répète cette réaction quatre fois en parallèle, avec les quatre didésoxyribonucléotides différents.
Par exemple, dans la réaction où on a ajouté du ddGTP, la synthèse s'arrête au niveau des G. Le mélange réactionnel contenant, à la fois du dGTP et un peu de ddGTP, la terminaison se fait de manière statistique suivant que l'ADN polymérase utilise l'un ou l'autre de ces nucléotides. Il en résulte un mélange de fragments d’ADN de tailles croissantes, qui se terminent tous au niveau d'un des G dans la séquence. Ces fragments sont ensuite séparés par électrophorèse sur un gel de polyacrylamide[6], ce qui permet ainsi de repérer la position des G dans la séquence.
La détection des fragments ainsi synthétisés se fait en incorporant un traceur dans l'ADN synthétisé. Initialement ce traceur était radioactif ; aujourd'hui, on utilise des traceurs fluorescents, attachés soit à l'oligonucléotide, soit au didésoxyribonucléotide.
Méthode de Maxam et Gilbert
Cette méthode est basée sur une dégradation chimique de l'ADN et utilise les réactivités différentes des quatre bases A, T, G et C, pour réaliser des coupures sélectives[7]. En reconstituant l'ordre des coupures, on peut remonter à la séquence des nucléotides de l'ADN correspondant. On peut décomposer ce séquençage chimique en six étapes successives :
- Marquage : Les extrémités des deux brins d'ADN à séquencer sont marquées par un traceur radioactif (32P). Cette réaction s'effectue en général au moyen d'ATP radioactif et de polynucléotide kinase.
- Isolement du fragment d'ADN à séquencer. Celui-ci est séparé au moyen d'une électrophorèse sur un gel de polyacrylamide. Le fragment d'ADN est découpé du gel et récupéré par diffusion.
- Séparation de brins. Les deux brins de chaque fragment d'ADN sont séparés par dénaturation thermique, puis purifiés par une nouvelle électrophorèse.
- Modifications chimiques spécifiques. Les ADN simple-brin sont soumis à des réactions chimiques spécifiques des différents types de base. Walter Gilbert a mis au point plusieurs types de réactions spécifiques, effectuées en parallèle sur une fraction de chaque brin d'ADN marqué : par exemple, une réaction pour les G (alkylation par le sulfate de diméthyle), une réaction pour les G et les A (dépurination), une réaction pour les C, ainsi qu'une réaction pour les C et les T (hydrolyse alcaline). Ces différentes réactions sont effectuées dans des conditions très ménagées, de sorte qu'en moyenne chaque molécule d'ADN ne porte que zéro ou une modification.
- Coupure. Après ces réactions, l'ADN est clivé au niveau de la modification par réaction avec une base, la pipéridine.
- Analyse. Pour chaque fragment, les produits des différentes réactions sont séparés par électrophorèse en conditions dénaturantes et analysés pour reconstituer la séquence de l'ADN. Cette analyse est analogue à celle que l'on effectue pour la méthode de Sanger.
Séquençage de génome entier
La connaissance de la structure d'un génome dans son entièreté peut passer par son séquençage. Cependant, la taille des génomes étant de plusieurs millions de bases (ou mégabases), il est nécessaire de coupler les approches de biologie moléculaire avec celle de l'informatique pour pouvoir traiter un nombre aussi important de données.
Deux grands principes de séquençage de génome entier sont utilisés. Dans les deux cas, l'ADN génomique est préalablement fragmenté par des méthodes enzymatiques (enzymes de restriction) ou physiques (ultrasons) :
- la méthode de séquençage par ordonnancement hiérarchique consiste à classer les fragments génomiques obtenus avant de les séquencer ;
- la méthode globale (ou whole-genome shotgun) ne fait pas de classement des fragments génomiques obtenus mais les séquence dans un ordre aléatoire. Une analyse bio-informatique faisant appel à un assembleur permet ensuite de réordonner les fragments génomiques par chevauchement des séquences communes.
La principale différence entre ces deux principes est que l'ordonnancement hiérarchique essaie d'aligner un jeu de clones de grande taille (~ 100 kb) alors que dans la méthode globale le génome entier est réduit en fragments de petite taille qui sont séquencés puis alignés.
Ordonnancement hiérarchique
Après extraction, l'ADN génomique est découpé par sonication en fragments de 50 à 200 kb puis cloné dans un vecteur adapté comme les chromosomes artificiels bactériens ou BAC. Le nombre de clones doit permettre une couverture de 5 à 10 fois la longueur totale du génome étudié. Le chevauchement et l'ordonnancement des clones est réalisé soit par hybridation de sondes spécifiques, soit par analyse des profils de restriction, soit plus fréquemment par un ordonnancement après séquençage et hybridation des extrémités des BAC. Après ordonnancement des clones, ils sont fragmentés et séquencés individuellement, puis assemblés par alignement bio-informatique.
Les avantages de cette méthode sont une plus grande facilité d'assemblage des fragments grâce aux chevauchement des BAC, la possibilité de comparer les fragments aux banques de données disponibles, et la possibilité de partager le travail de séquençage entre plusieurs laboratoires, chacun ayant en charge une région chromosomique.
L'inconvénient majeur est la difficulté de cloner des fragments contenant des séquences répétées très fréquentes dans certains génomes, comme ceux des mammifères, ce qui rend difficile l'analyse bio-informatique finale.
Méthode globale ou Shotgun
Il s'agit d'une méthode de séquençage d'ADN génomique initialement imaginée dans le laboratoire de Frederick Sanger à Cambridge à la fin des années 1970 pour séquencer les premiers génomes de virus[8].
Cette méthode a été popularisée par Craig Venter pour le séquençage des grands génomes, en particulier au sein de la société Celera Genomics. La première application fut le séquençage de génomes bactériens, puis du génome de la drosophile et enfin du génome humain et murin. Pour réaliser un séquençage de génome complet à l'aide de cette technique, deux à trois banques composées de fragments aléatoires d'ADN génomique sont réalisées. Entre les banques, les fragments divergent aussi bien en taille qu'en localisation sur le génome. À partir de ces banques, de nombreux clones sont séquencés puis assemblés. La séquence totale est obtenue en traitant l'ensemble des banques à l'aide d'outils bio-informatiques, en alignant les fragments à l'aide des séquences chevauchantes.
Les avantages par rapport au séquençage par ordonnancement hiérarchique sont la rapidité de la technique et un coût plus faible. L'inconvénient est que le traitement informatique ne permet pas d'aligner des fragments comportant des séquences répétées de grande taille qui sont fréquemment présentes dans les génomes des mammifères.
Cette méthode est couramment désignée sous le nom de shotgun (fusil à canon scié), ou encore Whole Genome Shotgun (WGS). Cette métaphore illustre le caractère aléatoire de la fragmentation initiale de l'ADN génomique : on arrose tout le génome, un peu comme se dispersent les plombs de ce type d'arme à feu.
Autres méthodes
Séquençage par hybridation
Le séquençage par hybridation repose sur l’utilisation de puces à ADN contenant de plusieurs centaines (pour les puces de première génération) à plusieurs milliers d’oligonucléotides. L’ADN à analyser est coupé en de multiples fragments qui sont ensuite incubés sur la puce où ils vont s’hybrider avec les oligonucléotides dont ils sont complémentaires. La lecture de la puce (la détection des oligonucléotides hybridés), permet d’obtenir le spectre de la séquence d’ADN, c’est-à-dire sa composition en sous-séquences de n nucléotides, où n est la taille des sondes sur la puce utilisée. Le traitement informatique du spectre permet ensuite de reconstituer la séquence entière[9].
Séquençage haut débit (HTS)
une adaptation de la technique de Sanger qui utilise la fluorescence à la place de la radioactivité. Les didésoxynucléotides incorporés sont marqués spécifiquement par des molécules fluorescentes ou fluorophores « fluorochromes » (ddATP-JOE, ddCTP-5-FAM, ddGTP-TAMRA et ddTTP-ROX).
La réaction de séquence est réalisée par PCR. La Taq polymerase effectue l’élongation jusqu’à l’incorporation d’un didésoxynucléotide marqué par fluorescence. Les fragments synthétisés sont ensuite séparés par électrophorèse.
Un automate prélève la réaction de séquence et l’injecte dans un capillaire contenant un polymère de polyacrylamide. Lors de la migration, un système optique laser détecte la fluorescence passant devant la fenêtre du laser et qui est émise par le ddNTP terminateur du fragment sous l’excitation (lumière verte pour JOE « ddATP », bleue pour 5-FAM « ddCTP », jaune pour TAMRA « ddGTP » et rouge pour ROX « ddTTP ».
En séparant ces molécules par électrophorèse en fonction de leur taille, on peut lire les lettres successives qui apparaissent sous forme de courbes sur un électrophorégramme (ou fluorogramme) dont la fluorescence correspond à la base de ce ddNTP terminateur. Un logiciel d’analyse permet de faire la correspondance entre les courbes de fluorescence et le nucléotide incorporé.
L’information fait l’objet d’un enregistrement électronique, et la séquence interprétée est stockée dans la banque de données de l’ordinateur. Ce type de séquençage est dit à haut débit car de nombreuses séquences peuvent être réalisées dans le même temps. En effet, selon les modèles de séquenceur, 1, 6, 12 voire 36 capillaires peuvent fonctionner en parallèle, sachant que l’automate peut injecter successivement 96 réactions de séquences, contenues dans une plaque, dans chacun des capillaires. La longueur de lecture est d’environ 1kb par séquence. Le temps d’un passage (« run ») d’une séquence est d’environ 10 minutes. En une nuit, avec 12 capillaires, le séquenceur permet d’obtenir de manière automatique la lecture d’1 Mb.
Comparaison des méthodes de séquençage nouvelle-génération[10],[11]
| Méthode | Longueur de la lecture | précision | Lecture par expérience | temps d'expérience | coût par 1 million de bases (en dollar américain $) | Avantages | Inconvénients |
|---|---|---|---|---|---|---|---|
| Séquençage d'une seule molécule en temps réel (Pacific Biosciences) | 10,000 pb à 15,000 pb en moyenne (14,000 pb N50); longueur de lecture maximale >40,000 bases[12],[13],[14] | 87%[15] | 50,000 par cellule, ou 500–1000 megabases[16],[17] | 30 minutes à 4 heures[18] | $0.13–$0.60 | lectures longues. Rapide. Détecte 4mC, 5mC, 6mA[19] | débit modéré, l'équipement peut être très coûteux |
| Ion semiconductor (Séquençage Ion Torrent) | jusqu'à 400 pb | 98 % | jusqu'à 80 millions | 2 heures | $1 | l'équipement le moins cher, rapide | erreurs d'homopolymères |
| Pyroséquençage (454) | 700 pb | 99,9 % | 1 million | 24 heures | $10 | lectures longues, rapide | l'expérience coûte cher, erreurs d'homopolymères |
| Séquençage par synthèse (Illumina) | 50 à 300 pb | 99,9 % | jusqu'à 6 milliards | 1 à 11 jours[20] | $0.05 à $0.15 | Potentiel de rendement élevé de séquence, selon le modèle de séquenceur et l'application souhaitée | L'équipement peut être très coûteux. Nécessite des concentrations élevées d'ADN. |
| Séquençage par ligation (séquençage SOLiD) | 50+35 ou 50+50 bp | 99,9 % | NA | 20 minutes à 3 heures | $2400 | lectures longues. Utile pour de nombreuses applications. | Plus cher et peu pratique pour les grands projets de séquençage. Cette méthode nécessite aussi du temps pour l'étape de clonage de plasmide ou PCR. |
.jpg.webp)

Nombre de machines mises en service par plate-forme[21]
| Nom | Nombre de machines (dans le monde) |
|---|---|
| Illumina HiSeq 2000 | 5490 |
| Illumina Genome Analyser 2x | 411 |
| Roche 454 | 382 |
| ABI SOLiD | 326 |
| Ion Torrent | 301 |
| Illumina MiSeq | 299 |
| Ion Proton | 104 |
| Pacific Biosciences | 50 |
| Oxford Nanopore MinION | 14 |
| Illumina NextSeq | 3 |
Technologies en développement : Nanopore
Le séquençage Nanopore est une méthode en cours de développement depuis 1995[22],[23] pour le séquençage d'ADN.
Un nanopore est simplement un petit trou d'un diamètre intérieur de l'ordre de 1 nanomètre. Certaines protéines cellulaires transmembranaires poreuses agissent comme des nanofils, des nanopores ont également été réalisés par gravure d'un trou légèrement plus grand (plusieurs dizaines de nanomètres) dans un morceau de silicium.
La théorie derrière le séquençage des nanopores est la suivante: lorsqu'un nanopore est immergé dans un fluide conducteur et qu'un potentiel (tension) est appliquée à travers lui, un courant électrique dû à la conduction des ions à travers le nanopore peut être observée. La quantité de courant est très sensible à la taille et la forme du nanopore. Si les nucléotides simples (bases), des brins d'ADN ou d'autres molécules passent à travers ou à proximité du nanopore, cela peut créer une variation caractéristique de l'amplitude du courant à travers le nanopore.
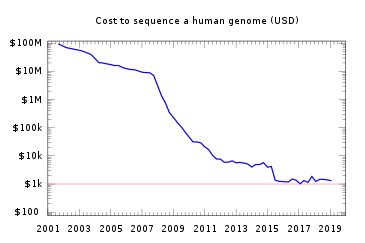
Baisse du prix de séquençage
Au début de la seconde moitié du XXe siècle, le rapport de l’être humain à la médecine était encore dominé par la volonté de comprendre et de soigner les maladies et différentes menaces pour l’organisme. Cependant, la compréhension du fonctionnement de celui-ci s’est largement approfondie ces dernières décennies, notamment grâce à l’amélioration et l’apparition de différentes techniques. Le concept même de santé, signifiant alors plutôt une absence de pathologie, a été naturellement redéfini pour désormais plutôt signifier une sensation de bien-être global d’un individu, à la fois physique et moral. Ainsi, de nouvelles stratégies commerciales se sont démocratisées pour proposer à chaque individu de prendre soin lui-même de son intégrité physique. (médicaments sans prescription médicale, nourriture saine, etc.)[24].
Le séquençage de l’ADN est une technique au cœur même de cette redéfinition de la conception de la santé et du rapport au “vivant” en général, puisqu’elle suggère un traitement optimal et personnalisé pour chacun. Le marché des données génétiques s’est très rapidement développé et de nombreux investissements depuis sa création ont permis une très forte baisse des prix.
Le premier séquençage complet d’un génome humain s’est achevé en 2003 et a nécessité une dizaine d'années de travail, avec un investissement total de 2,7 milliards de dollars[25]. À l’époque, la méthode de Sanger était encore massivement employée pour décrypter les quelque 3 milliards de paires de nucléotides qui composent notre ADN. De nombreux projets sont ensuite apparus, (notamment 1000 Génomes, ENCODE…) et de nouvelles machines (citées au-dessus) ont été développées avec pour objectif de générer la séquence complète d’un génome humain pour moins de 1000 dollars[26]. Avec l’amélioration des méthodes de séquençage, le prix du séquençage partiel d’un génome humain en haute qualité était estimé à 14 millions de dollars en 2006[27], relativement moins coûteux par rapport au projet achevé en 2003. Fin 2015, le prix pour générer une même séquence était de 1500 dollars environ.
Avec l’apparition de ces nouvelles méthodes bien plus efficaces, regroupées sous l’acronyme des NGS, plus rapides et moins couteuses, le marché du séquençage de l’ADN a explosé et de nombreuses applications dans des domaines variés sont aujourd’hui disponibles. Certaines sociétés comme Illumina proposent maintenant un service de séquençage d'ADN, financièrement accessible pour les particuliers[28].
Applications
Le séquençage de l’ADN peut être utilisé pour déterminer la séquence de gènes individuels, de grandes régions génétiques, des chromosomes complets ou des génomes entiers, de n'importe quel organisme. Le séquençage de l'ADN est devenu une technologie clé dans de nombreux domaines de la biologie et d'autres sciences telles que la médecine, la médecine légale ou l'anthropologie.
Biologie moléculaire
En biologie moléculaire, le séquençage du génome permet l’étude des protéines encodées, les chercheurs identifient les changements dans les gènes et les associent avec certaines maladies afin de cibler de potentiels médicaments.
Le séquençage a permis d’appréhender l’origine génétique de certains cancers qui surviennent en raison de l'accumulation de mutations dans des gènes critiques qui modifient les programmes normaux de prolifération, de différenciation et de décès cellulaire. La kinase RAS-RAF-MEK-ERK-MAP fait appel aux réponses cellulaires aux signaux de croissance et dans environ 15 % du cancer humain le gène RAS est muté provoquant une forme oncogène[29].
Biologie évolutive
Étant donné que l'ADN est une macromolécule informative en termes de transmission d'une génération à l'autre, le séquençage de l'ADN est utilisé en biologie évolutive pour étudier la manière dont les différents organismes sont liés et comment ils ont évolué, basée sur des études collaboratives entre paléogénétitiens et anthropologues. L’analyse de l’ADN de tissus humains principalement osseux et dentaires, inhumés dans des nécropoles, permet de définir des haplogroupes et d’estimer leur origine biogéographique ainsi que les voies de migration qu’ils ont pu emprunter il y a des centaines ou milliers d’années, de comparer leurs caractéristiques génétiques avec celles des populations actuelles, ou encore d’établir certains de leurs traits physiques[30]. En raison de la baisse du prix du séquençage du génome, des entreprises proposent au public, en service payant, de retracer les origines d'une personne à partir d’un simple kit à utiliser chez soi.
Génétique médicale
Les généticiens médicaux peuvent séquencer les gènes des patients pour déterminer s'il existe un risque de maladies génétiques. Il s'agit d'un examen des caractéristiques génétiques de la personne. Le diagnostic est classiquement pré ou post-natal. Par exemple, le diagnostic prénatal peut détecter une maladie héréditaire responsable d'un handicap grave ou de troubles comportementaux et psychiques et donner le choix aux parents dont l'enfant est diagnostiqué de poursuivre ou non la grossesse. L’information sur les variations génétiques (single nucleotide polymorphisms) guide également la prise en charge thérapeutique et permet de faire du conseil génétique pour les personnes de la famille.
De plus en plus, l'examen des caractéristiques génétiques se fait par séquençage à haut débit de l'ADN (NGS). En général à l'heure actuelle, on séquence plutôt seulement les parties codantes des gènes, dans lesquels les 2/3 des mutations sont décrites. Le NGS permet donc de séquencer en une seule fois l'ensemble des parties codantes des gènes d'une personne, ce que l'on appelle un exome.
En diagnostic prénatal, le DPNI est en train de s'implanter comme une technique de dépistage précoce et sans danger de la trisomie 21 ou d'autres anomalies chromosomiques, ou même de certaines mutations ponctuelles. Ce n'est pas un diagnostic, mais seulement un dépistage. Il consiste à prélever du sang à la maman pendant la grossesse. Ce sang contient naturellement une petite quantité de fragments d'ADN du fœtus, et les généticiens ne peuvent pas le séparer des fragments d'ADN appartenant à la mère, que l'on peut également trouver dans le sang. Le DPNI est donc un séquençage haut débit de tous les fragments d'ADN circulant dans le sang maternel, puis une analyse informatique des résultats. DPNI signifie Dépistage Prénatal par technique Non-Invasive. En fonction des résultats, une confirmation de l'anomalie est indiquée, qui implique une amniocentèse[31].
Médecine reproductive
La médecine de la reproduction est la branche de la médecine étudiant la physiologie de la reproduction ainsi que sa pathologie, l'infertilité. Cette approche de la médecine vise à améliorer la santé de la reproduction.
Le séquençage de l’ADN, notamment des cellules sexuelles, a permis d’appréhender le modifications génétiques causant un dérèglement de la fertilité. De futurs traitements génétiques sont envisagés visant à se prémunir des maladies héréditaires, par exemple la trisomie 21 est due à la non-expression d’un gène responsable de l'inactivation du chromosome X lors de la fécondation[32]. Cependant, des questions bioéthiques se posent sur le traitement de l’ADN pour la procréation.
Microbiologie médicale
Le séquençage à haut débit a également fait son entrée dans le domaine de la microbiologie médicale[33]. En bactériologie par exemple, même si on peut retrouver la même espèce bactérienne (ex : Staphylocoque doré) dans deux prélèvements de patients différents, il ne s'agit pas forcément d'une transmission directe de patient à patient. En effet, sous la même espèce bactérienne sont regroupées de nombreuses souches très différentes et possèdent ainsi des génomes différents. Le séquençage du génome entier permet par exemple de déterminer à quel point ces génomes sont différents en quantifiant le nombre de mutations (SNPs) entre les organismes. Lors de transmission directe d'une bactérie d'un patient à un autre, le nombre de mutation de différence est donc très faible.
Globalement, le séquençage à haut débit de génomes bactériens entiers peut être utile pour :
- Investiguer des épidémies et confirmer une transmission (directe de patient à patient[34] ou d'une source environnementale commune à des patients[35]).
- Détecter des gènes de résistance aux antibiotiques et / ou des mutations dans des gènes cibles des antibiotiques qui confèrent des résistances[36].
- Détecter des gènes codant des facteurs de virulence (en détectant le pathotype de Escherichia coli par exemple[37]).
- Développer ou perfectionner de nouveaux outils de diagnostic moléculaire (ex : PCR)[38].
Médecine légale
L’ADN d’une personne peut être transféré par contact sur des objets ou sur des personnes. Cet ADN provient des cellules issues de différentes matrices, le sang, le sperme, les éléments pileux, les cellules épithéliales[39]. (Le séquençage de l'ADN peut être utilisé avec des méthodes de profilage de l'ADN pour l'identification médico-légale et les tests de paternité. Il faut toutefois préciser qu'un test de paternité n'a de valeur juridique en France que si c'est un juge qui l'a ordonné.
Notes et références
- (en) F. Sanger, G.M. Air, B.G. Barrell, N.L. Brown, A.R. Coulson, C.A. Fiddes, C.A. Hutchison, P.M. Slocombe et M. Smith, « Nucleotide sequence of bacteriophage phi X174 DNA », Nature, vol. 265, , p. 687-695 (PMID 870828)
- (en) Joachim Messing, « New M13 vectors for cloning », Methods Enzymol., vol. 101, , p. 20-78 (PMID 6310323)
- (en) L.M. Smith, J.Z. Sanders, R.J. Kaiser, P. Hughes, C. Dodd, C.R. Connell, C. Heiner, S.B. Kent et L.E. Hood, « Fluorescence detection in automated DNA sequence analysis », Nature, vol. 321, , p. 674-679 (PMID 3713851)
- (en) H. Swerdlow, J.Z. Zhang, D.Y. Chen, H.R. Harke, R. Grey, S.L. Wu, N.J. Dovichi et C. Fuller, « Three DNA sequencing methods using capillary gel electrophoresis and laser-induced fluorescence », Anal. Chem., vol. 63, , p. 2835-2841 (PMID 1789449)
- (en) F. Sanger, S. Nicklen et A.R. Coulson, « DNA sequencing with chain-terminating inhibitors », Proc. Natl. Acad. Sci. USA, vol. 74, , p. 5463-5467 (PMID 271968)
- (en) F. Sanger et A.R. Coulson, « The use of thin acrylamide gels for DNA sequencing », FEBS Lett., vol. 87, , p. 107-110 (PMID 631324)
- (en) Allan M. Maxam et Walter Gilbert, « A new method for sequencing DNA », Proc. Natl. Acad. Sci. USA, vol. 74, , p. 560-564 (PMID 265521)
- (en) Roger Staden, « A strategy of DNA sequencing employing computer programs. », Nucleic Acids Res., vol. 6', , p. 2601-2610 (PMID 461197)
- (en) Ji-Hong Zhang, Ling-Yun Wu et Xiang-Sun Zhang, « Reconstruction of DNA sequencing by hybridization », Bioinformatics, vol. 19, no 1, , p. 14–21 (PMID 12499288, lire en ligne [PDF])
- Michael A Quail, Miriam Smith, Paul Coupland et Thomas D Otto, « A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers », BMC Genomics, vol. 13, , p. 341 (ISSN 1471-2164, PMID 22827831, PMCID 3431227, DOI 10.1186/1471-2164-13-341, lire en ligne, consulté le )
- Lin Liu, Yinhu Li, Siliang Li et Ni Hu, « Comparison of next-generation sequencing systems », Journal of Biomedicine & Biotechnology, vol. 2012, , p. 251364 (ISSN 1110-7251, PMID 22829749, PMCID 3398667, DOI 10.1155/2012/251364, lire en ligne, consulté le )
- « New Products: PacBio's RS II; Cufflinks », sur GenomeWeb (consulté le )
- « After a Year of Testing, Two Early PacBio Customers Expect More Routine Use of RS Sequencer in 2012 », sur GenomeWeb (consulté le )
- (en-US) « Pacific Biosciences Introduces New Chemistry With Longer Read Lengths to Detect Novel Features in DNA Sequence and Advance Genome Studies of Large Organisms », sur GlobeNewswire News Room (consulté le )
- Chen-Shan Chin, David H. Alexander, Patrick Marks et Aaron A. Klammer, « Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data », Nature Methods, vol. 10, , p. 563-569 (ISSN 1548-7105, PMID 23644548, DOI 10.1038/nmeth.2474, lire en ligne, consulté le )
- « De novo bacterial genome assembly: a solved problem? » (consulté le )
- David A. Rasko, Dale R. Webster, Jason W. Sahl et Ali Bashir, « Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany », The New England Journal of Medicine, vol. 365, , p. 709-717 (ISSN 1533-4406, PMID 21793740, PMCID 3168948, DOI 10.1056/NEJMoa1106920, lire en ligne, consulté le )
- Ben Tran, Andrew M. K. Brown, Philippe L. Bedard et Eric Winquist, « Feasibility of real time next generation sequencing of cancer genes linked to drug response: results from a clinical trial », International Journal of Cancer. Journal International Du Cancer, vol. 132, , p. 1547-1555 (ISSN 1097-0215, PMID 22948899, DOI 10.1002/ijc.27817, lire en ligne, consulté le )
- Iain A. Murray, Tyson A. Clark, Richard D. Morgan et Matthew Boitano, « The methylomes of six bacteria », Nucleic Acids Research, vol. 40, , p. 11450-11462 (ISSN 0305-1048, PMID 23034806, PMCID 3526280, DOI 10.1093/nar/gks891, lire en ligne, consulté le )
- Arnoud H. M. van Vliet, « Next generation sequencing of microbial transcriptomes: challenges and opportunities », FEMS microbiology letters, vol. 302, , p. 1-7 (ISSN 1574-6968, PMID 19735299, DOI 10.1111/j.1574-6968.2009.01767.x, lire en ligne, consulté le )
- « High-throughput sequencing maps », sur omicsmaps.com (consulté le )
- George Church, David W. Deamer, Daniel Branton et Richard Baldarelli, United States Patent: 5795782 - Characterization of individual polymer molecules based on monomer-interface interactions, (lire en ligne)
- (en) John J. Kasianowicz, Eric Brandin, Daniel Branton et David W. Deamer, « Characterization of individual polynucleotide molecules using a membrane channel », Proceedings of the National Academy of Sciences, vol. 93, , p. 13770–13773 (ISSN 0027-8424 et 1091-6490, PMID 8943010, PMCID 19421, lire en ligne, consulté le )
- (en) Nikolas Rose, « The Politics of Life Itself », Theory, Culture & Society, (lire en ligne)
- Alexandre Léchenet, « Business, éthique, légalité... Le séquençage de l'ADN en questions », Le Monde, (lire en ligne).
- Jacques Hallard, « Génomique environnementale », Les Cahiers Prospectives CNRS, , p. 1-13 (lire en ligne, consulté le )
- https://www.genome.gov/sequencingcosts/
- « Sequencing | Sequencing Methods », sur www.illumina.com (consulté le )
- H. Davies, G. R. Bignell et C. Cox, « Mutations of the BRAF gene in human cancer », Nature, vol. 417, (lire en ligne)
- Froment, A. La génétique anthropologique entre archéologie et identité. Une archéologie pour le développement, 91. (lire en ligne)
- « La Génétique pour les Nuls - Patrice BOURGEOIS , Tara RODDEN-ROBINSON », sur www.pourlesnuls.fr (consulté le )
- J. Money, « Human behavior cytogenetics: Review of psychopathology in three syndromes—47, XXY; 47, XYY; and 45, X », Journal of Sex Research, vol. 11, (lire en ligne)
- Xavier Didelot, Rory Bowden, Daniel J. Wilson et Tim E. A. Peto, « Transforming clinical microbiology with bacterial genome sequencing », Nature Reviews. Genetics, vol. 13, no 9, , p. 601–612 (ISSN 1471-0064, PMID 22868263, PMCID PMC5049685, DOI 10.1038/nrg3226, lire en ligne, consulté le )
- Claudio U. Köser, Matthew T. G. Holden, Matthew J. Ellington et Edward J. P. Cartwright, « Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak », The New England Journal of Medicine, vol. 366, no 24, , p. 2267–2275 (ISSN 1533-4406, PMID 22693998, PMCID PMC3715836, DOI 10.1056/NEJMoa1109910, lire en ligne, consulté le )
- Jakko van Ingen, Thomas A. Kohl, Katharina Kranzer et Barbara Hasse, « Global outbreak of severe Mycobacterium chimaera disease after cardiac surgery: a molecular epidemiological study », The Lancet. Infectious Diseases, vol. 17, no 10, , p. 1033–1041 (ISSN 1474-4457, PMID 28711585, DOI 10.1016/S1473-3099(17)30324-9, lire en ligne, consulté le )
- M. J. Ellington, O. Ekelund, F. M. Aarestrup et R. Canton, « The role of whole genome sequencing in antimicrobial susceptibility testing of bacteria: report from the EUCAST Subcommittee », Clinical Microbiology and Infection: The Official Publication of the European Society of Clinical Microbiology and Infectious Diseases, vol. 23, no 1, , p. 2–22 (ISSN 1469-0691, PMID 27890457, DOI 10.1016/j.cmi.2016.11.012, lire en ligne, consulté le )
- Roy M. Robins-Browne, Kathryn E. Holt, Danielle J. Ingle et Dianna M. Hocking, « Are Escherichia coli Pathotypes Still Relevant in the Era of Whole-Genome Sequencing? », Frontiers in Cellular and Infection Microbiology, vol. 6, , p. 141 (ISSN 2235-2988, PMID 27917373, PMCID PMC5114240, DOI 10.3389/fcimb.2016.00141, lire en ligne, consulté le )
- S. M. Diene, C. Bertelli, T. Pillonel et N. Jacquier, « Comparative genomics of Neisseria meningitidis strains: new targets for molecular diagnostics », Clinical Microbiology and Infection: The Official Publication of the European Society of Clinical Microbiology and Infectious Diseases, vol. 22, no 6, , p. 568.e1–7 (ISSN 1469-0691, PMID 27085725, DOI 10.1016/j.cmi.2016.03.022, lire en ligne, consulté le )
- Christian Doutrepechui, « A. les empreintes génétique en pratique judiciaire », Bull. Acad. Natle Méd., (lire en ligne)
Voir aussi
Articles connexes
Liens externes
- Notices dans des dictionnaires ou encyclopédies généralistes :
- Questions à propos du séquençage du génome humain
- Un film d'animation pour comprendre: "Apport du séquençage de nouvelle génération dans le diagnostic des maladies rares"
 Portail de la biochimie
Portail de la biochimie  Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire